使用 HMMER 查找基因的同源序列
本文总阅读量 次
使用 HMMER 查找同源基因比 blast 更加准确,速度也更快,但使用方法很少有文章讲清楚,本文记录我使用 HMMER 的操作。
HMMER
HMMER 是基于隐马尔可夫模型,用于生物序列分析工作的一个非常强大的软件包,它的一般用途是识别同源蛋白或核苷酸序列和进行序列比对。与 BLAST、FASTA 等序列比对和数据库搜索工具相比,HMMER 更准确
从功能基因研究的角度来讲,相关的搜索,比如从序列数据库中,找同源的序列,或者对一个新的基因功能进行鉴定,使用 HMMER 比使用 blast 有着更高的灵敏度以及更高的搜索速度,但其应用远没有 blast 普及
所需工具
Linux 系统环境、HMMER 软件、pfam 网站(http://pfam-legacy.xfam.org/)
下载隐马尔科夫模型
我以拟南芥的 SBP 家族为例 打开 pfam,输入基因家族的 pf 号点击 go 进行查询
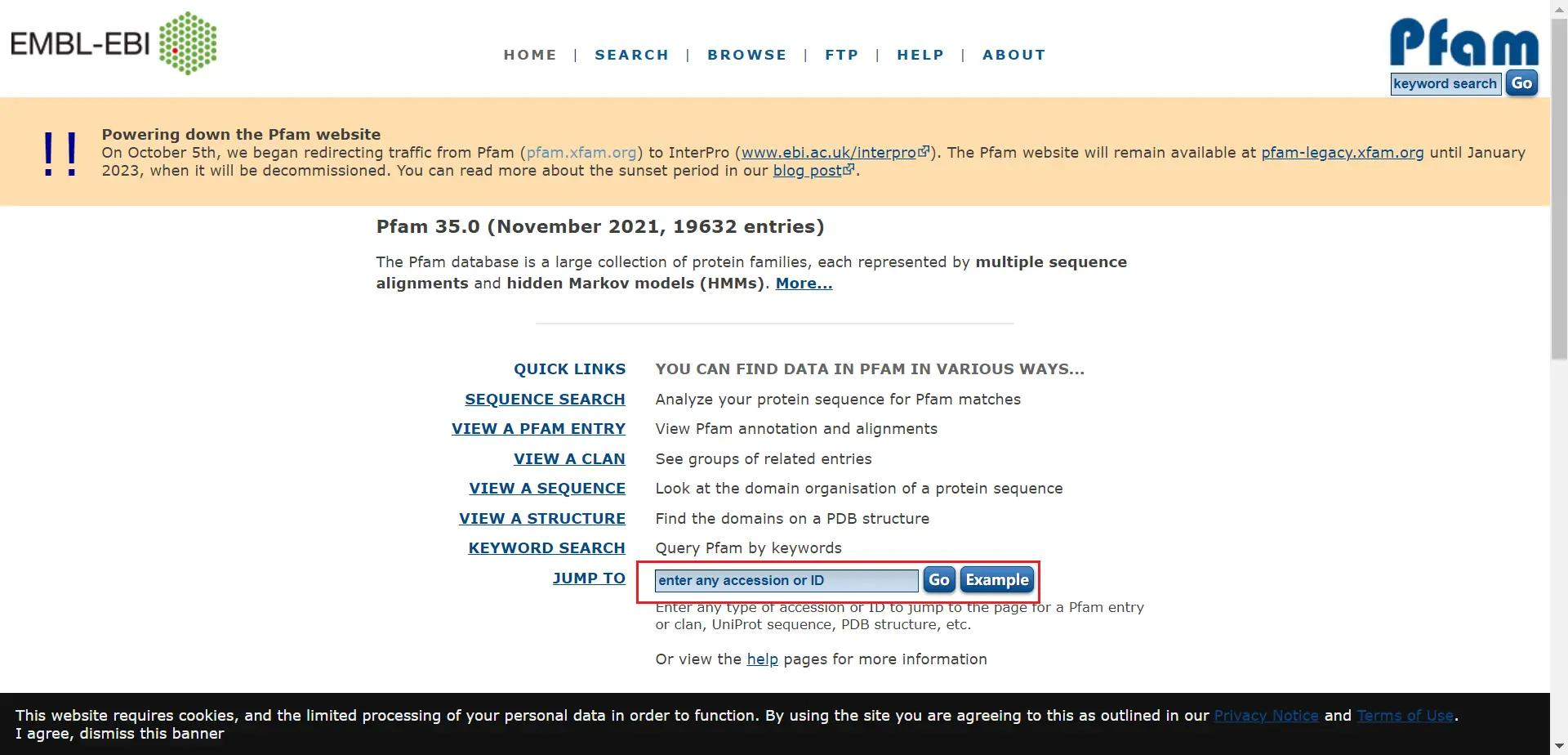
也可以通过查询关键字来查找
查询到之后,点击左边的 Curation & model 下载 hmm 文件

再点击 Alignments 选择 stockholm,点击 gengerate 后会自动下载一个 txt 文件

安装 HMMER
在 Linux 系统环境下,推荐使用 conda 安装 HMMER,conda 的安装配置教程请参照此文章 《Linux 下安装 conda》
#创建 Python3.7 的虚拟环境
conda install -n bioinfo python=3.7
#激活虚拟环境
conda activate bioinfo
#安装 hmmer
conda install hmmer
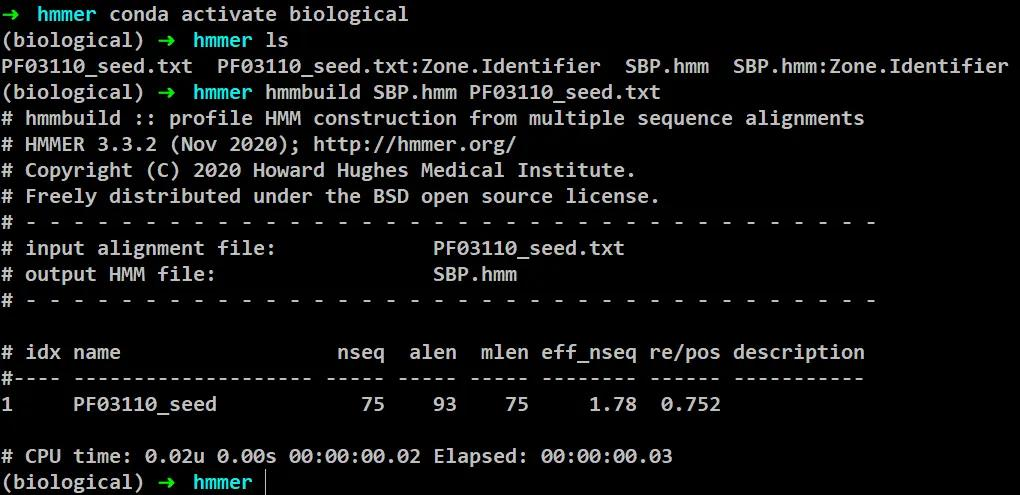
构建 hmm 模型
在工作目录下执行命令
hmmbuild [xxx.hmm] [xxx.txt] #构建 hmm 模型(使用我们再 pfam 网站下载的 hmm 文件和 txt 文件,要保证文件都在同一工作目录下
进行比对
在工作目录下执行命令
hmmsearch [hmm 文件] [待比对物种 fasta 文件] > [输出文件名]
#输出文件名以。out 结尾
#fasta 文件即整个物种的氨基酸序列 (fasta 文件名的缩写为 fa)
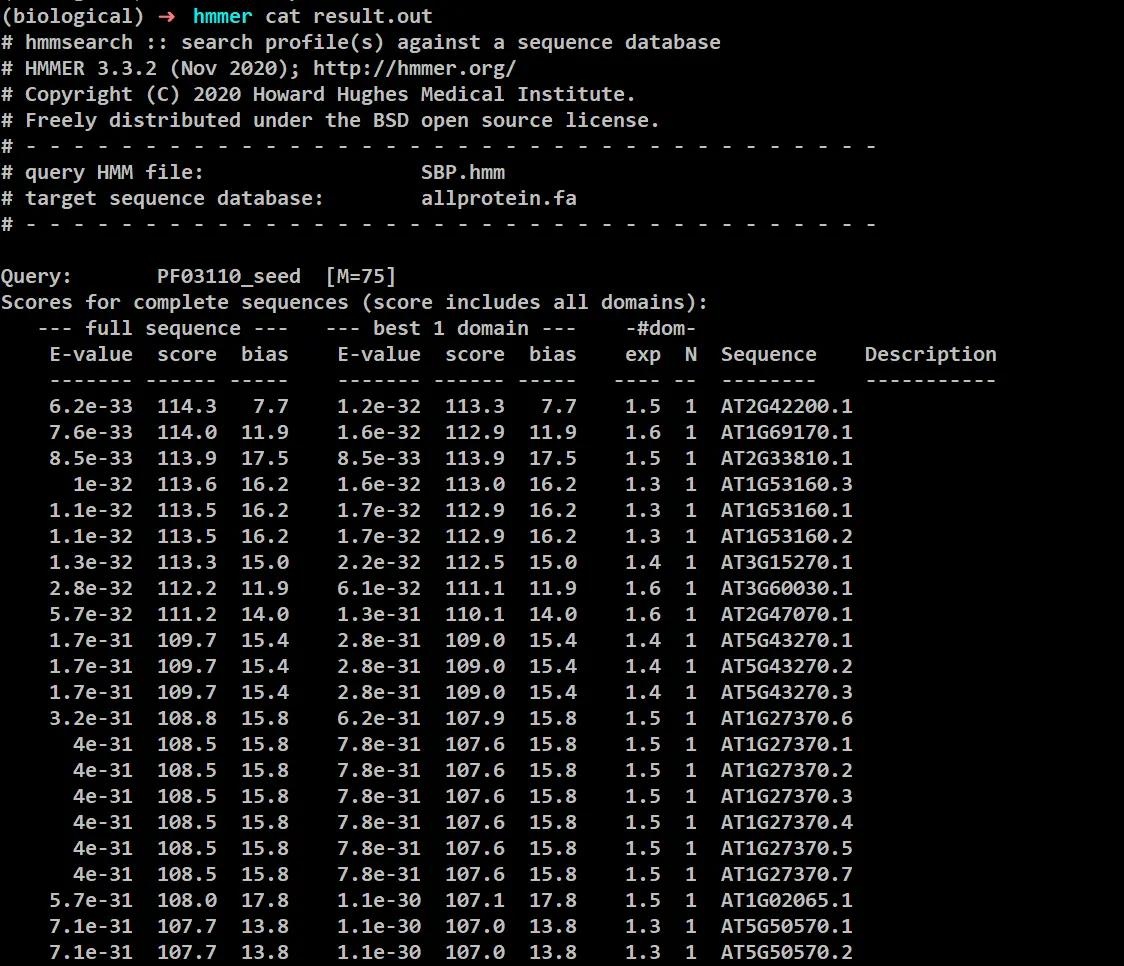
比对完成之后使用 cat 命令查看比对的结果
cat result.out
进一步筛选
随后我们可以通过各种数据库如 NCBI、TBtools 的比对工具等,进行进一步筛选,并用 MEME、CD-Search 进一步鉴定,排除“假阳性“。