专题:基因家族鉴定
本文总阅读量 次
完整的基因家族鉴定流程,包含基因家族鉴定和基因家族的性质分析、基因分析、进化分析等。
基因家族来源于同一个祖先,由同一个基因通过基因重复而产生两个或更多的拷贝而构成的一组基因,其在结构和功能上就有明显的相似性,编码相似的蛋白质产物,同一家族基因可以紧密排列在一起,形成一个基因簇,但多数时候,他们分散在同一染色体的不同位置,或存在于不同的染色体上,各自有不同的表达调控模式。
序列高度相似的序列,互为同源基因(homologous gene),归属于一个基因家族。
有时定义基因家族,从结构域角度来刻画。如:一类基因,其编码蛋白都含有同一个结构域,这一类基因是一个基因家族。这个定义信息更偏向功能信息,一般来说结构域决定某种功能,因为结构域序列保守,易形成稳定的三维结构。这与共同祖先的定义有些差别,很多结构域难找得到其共同祖先。另外一个基因的共同祖先定义比较复杂的,越是历史久远的祖先,因为物种的在进化过程中发生了很多丢失和增加事件。共同祖先是个相对的概念,比如植物的共同祖先,一般包括藻类及其它绿色植物,而被子植物共同祖先,根据已经测序的基因组,一般指单双子叶之前就可以。如果从共同祖先定义基因家族,很多已知的基因家族就要被分成很多个基因家族。有很多网站(数据库)专门收集结构域,比如 Pfam 和 InterPro,这两个数据库内容差不多。这些数据库以 Hmmer 算法为基础,根据 Uniprot 中包含的蛋白,进行序列连配找到保守的片段(结构域),再以这些序列使用 Hmmer 构建种子,保存这些种子。一个蛋白拿过来后,与这些种子比对,根据打分能判断出这个蛋白是不是含有这个结构域,这也是判断一个基因编码蛋白是不是属于这个家族。
研究思路
关于基因家族的鉴定思路主要包括以下部分:
基因家族成员鉴定
- 确定研究的基因家族
- 家族成员的基本特征确定(参考已有物种)
- 参考序列集合的准备或下载该基因家族的隐马尔科夫模型
- 目标物种序列和注释信息的下载或准备
- 双向 Blast 比对或 HMMER Search 获取可能的成员
- 基于保守结构域进行进一步筛选
- 结合系统发育树、保守结构域和保守基序(motif)等确定最终的基因家族成员
基因家族成员的性质分析
- 理化性质分析
- 染色体定位分析
基因家族成员的基因分析
- 基于 motif 分析成员序列保守特征与可视化(蛋白与核酸,可用于挖掘未知,尤其是核酸水平-非编码水平的保守)
- 基于 domain 分析成员结构域的保守型与可视化
- 基因结构的外显子分析和内含子分析
- 转录组的基因表达量分析
基因家族成员的进化分析
- 多序列比对与可视化
- 进化树构建与可视化
- 进化树+motif 分析+domain 分析+基因结构(外显子+内含子)
- 基因-共线性的定义与常见算法原理
- 物种内的共线性分析
- 不同物种之间的共线性分析
其它的分析
- 顺式作用元件分析
- GO 富集分析
- KEGG 分析
前期准备工作
查找基因的结构域
首先,当我们要以一个基因为起点,展开对其整个基因家族的 Bioinfo 分析,我们首先要确定它究竟属于哪一个基因家族,以水稻中的基因 Os05g0409300 为例,我们首先要收集这个基因的蛋白质序列,通过它的蛋白质序列来搜索它的结构域
打开 RAP-DB 网站首页的 ID converter 工具,输入基因 ID 进行转换,以 Os 开头的是 RAP ID,以 LOC 开头的是 MSU ID,这里我们要将 RAP ID 转为 LOC ID ,点击 ② 处的按钮选择 RAP ID 转为 LOC ID,点击 converter 进行转换
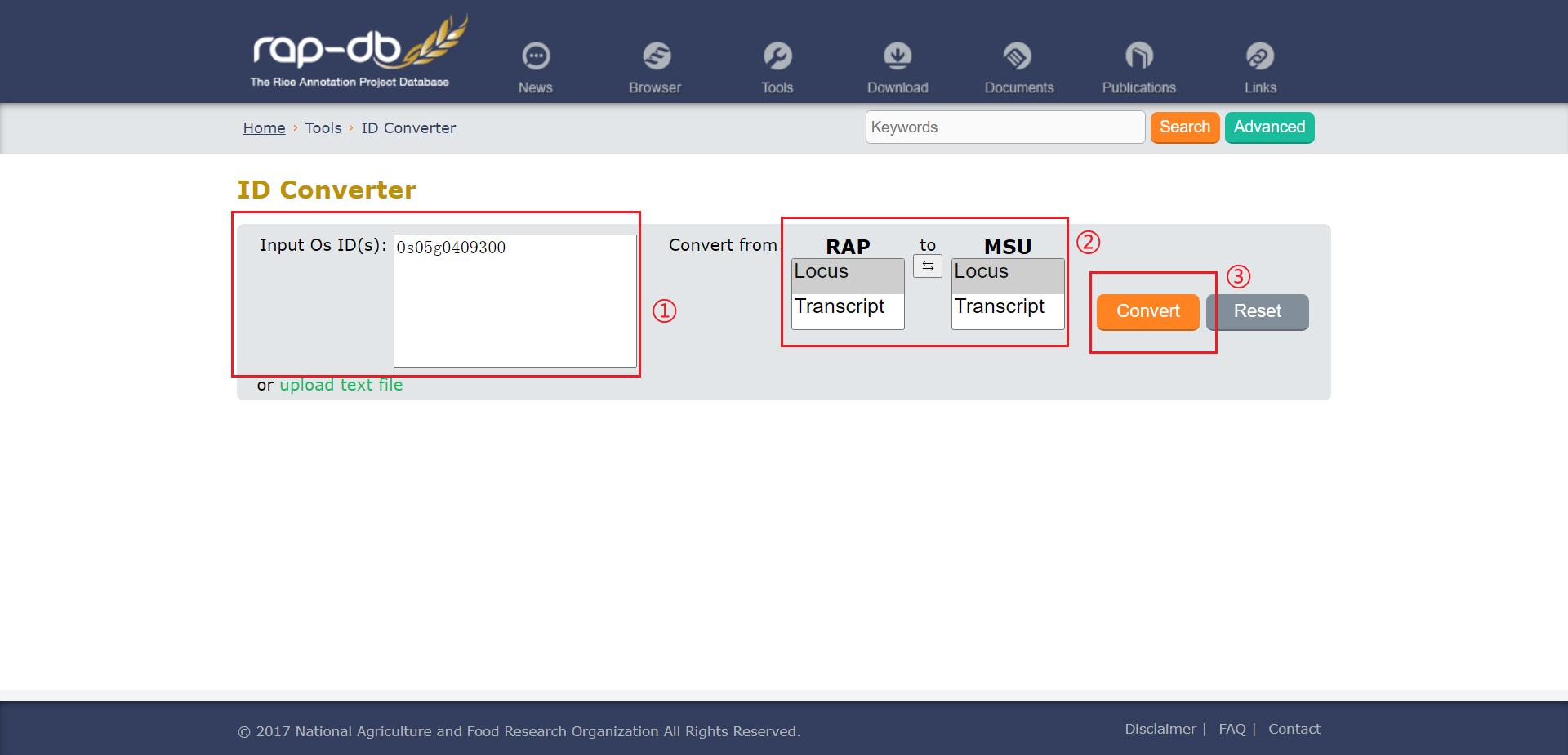
我们现在可以看到下方出现一个列表,点击 RAP ID 列的 Os05g0409300 进入该基因的详情页
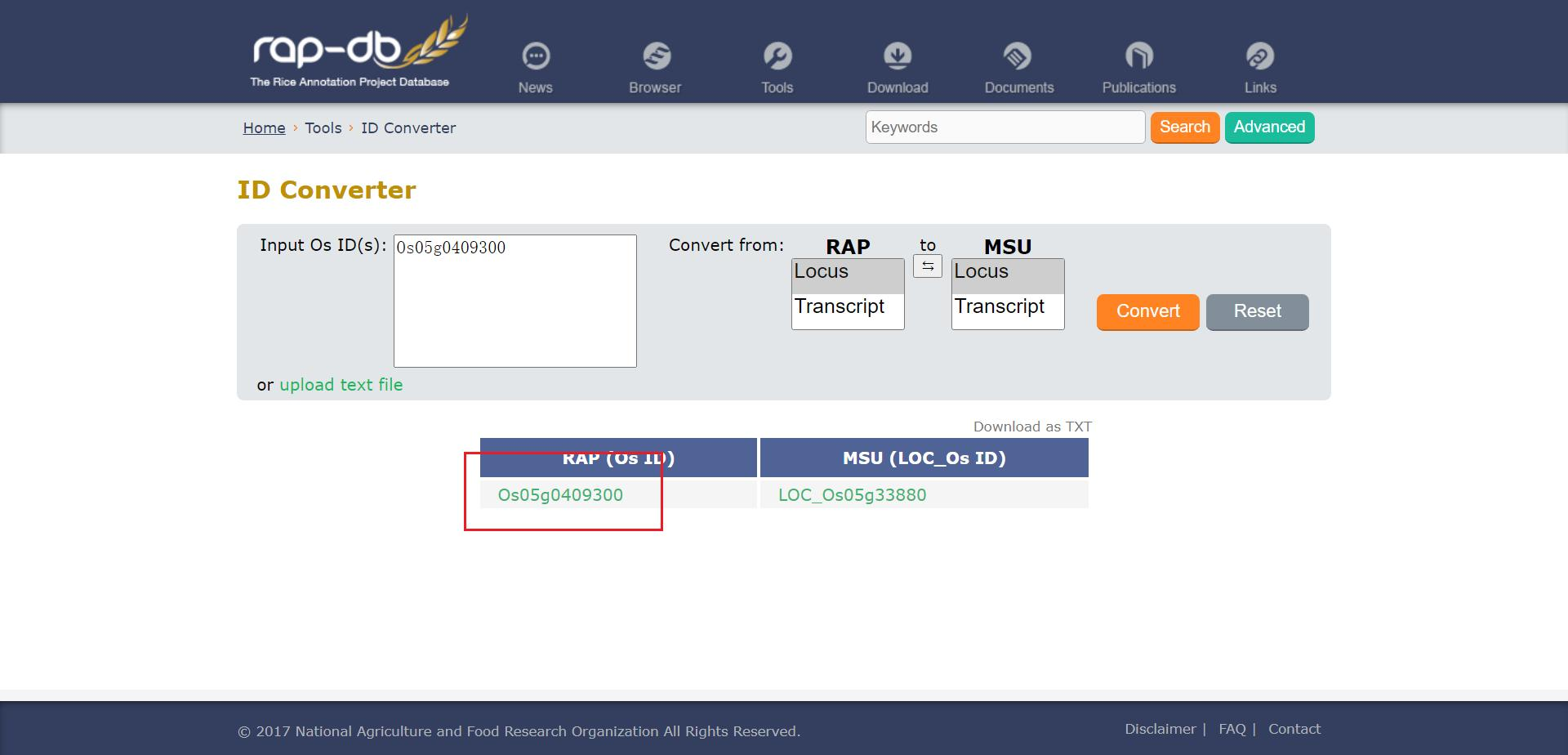
再点击 Transcript variants (转录变体)下的基因名称进入该基因转录本的详情页
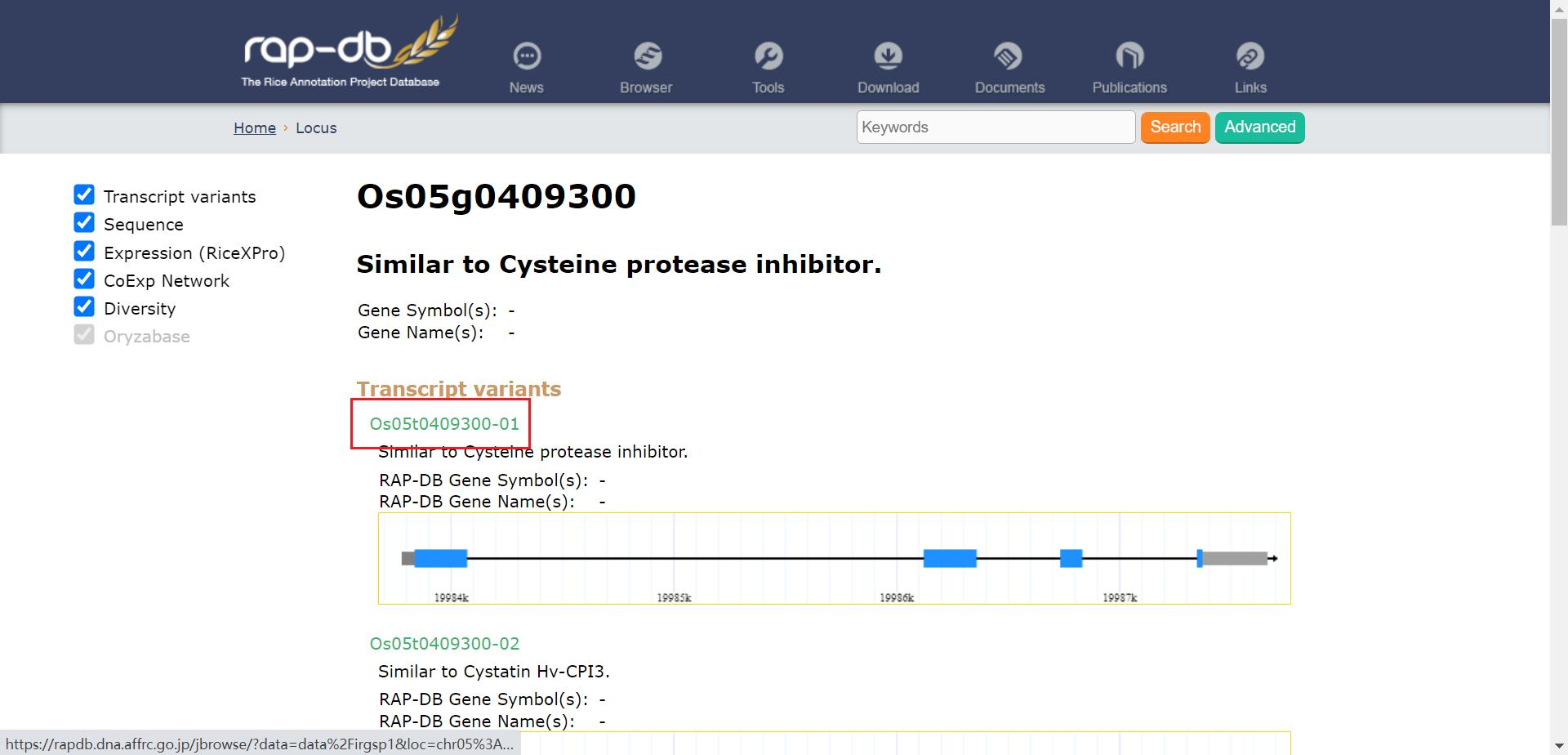
下滑到最下方即可得到蛋白质序列,复制下来保存即可

当然,也可以在 ID 转换后点击 LOC ID 列的基因名称,进入详情页后点击 Download Sequence 后下滑到最下方即可得到蛋白质序列,这两种方法得到的蛋白质序列是相同的,注意复制蛋白质序列时末尾的 * 不需要复制
随后打开 SMART 网站 粘贴刚刚得到的蛋白质序列搜索其结构域

搜索完成后我们可以看到,SMART 网站已经将结构域的位置等信息列出来了,下方表格中 E-value 小于十的负五次方的通常认为其可信,而 E-value 值为 N/A 的即为空值,不需要管它
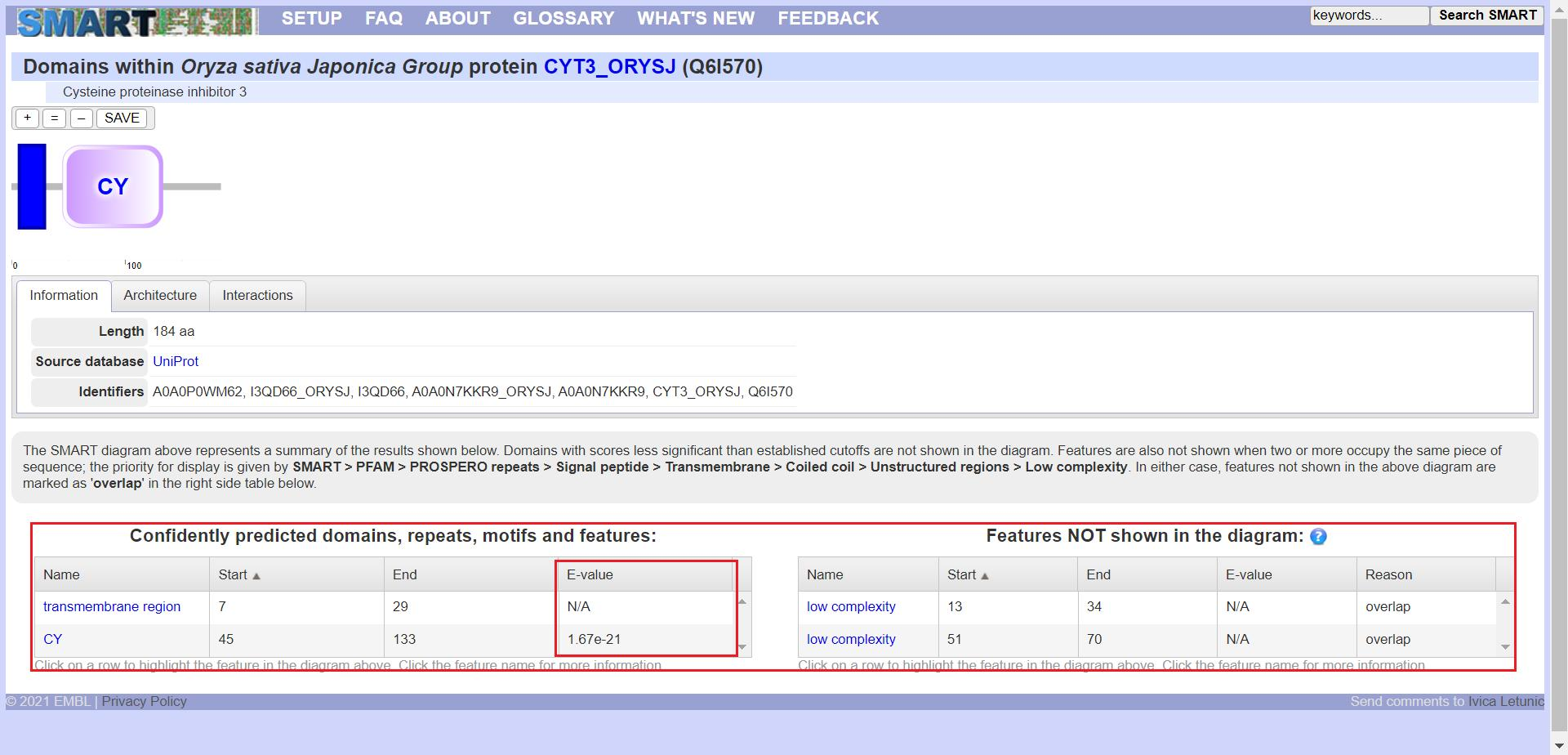
点击下方表格中的结构域名称我们可以得到该结构域的全称、描述等详细信息,可以看到我这个基因的结构域为 Cystatin,所以后面我便以水稻 Cystatin 基因家族为基础展开一系列生物信息学分析

搜索相关文献
通过 NCBI、知网、web of science 等文献检索网站查找结构域相关的文献,看该结构域是否已经被鉴定以及关于此结构域的研究成功有哪些,我查到一篇文献已经对其进行了生物信息学鉴定,并且将该家族基因命名为 OsCY-1 的形式,所以后面我也沿用该文献的命名对其进行研究
基因组序列文件与注释文件下载
首先使用 Ensembl Plants 网站下载物种的全部基因组序列作为参考序列,并下载 Gff3 注释文件
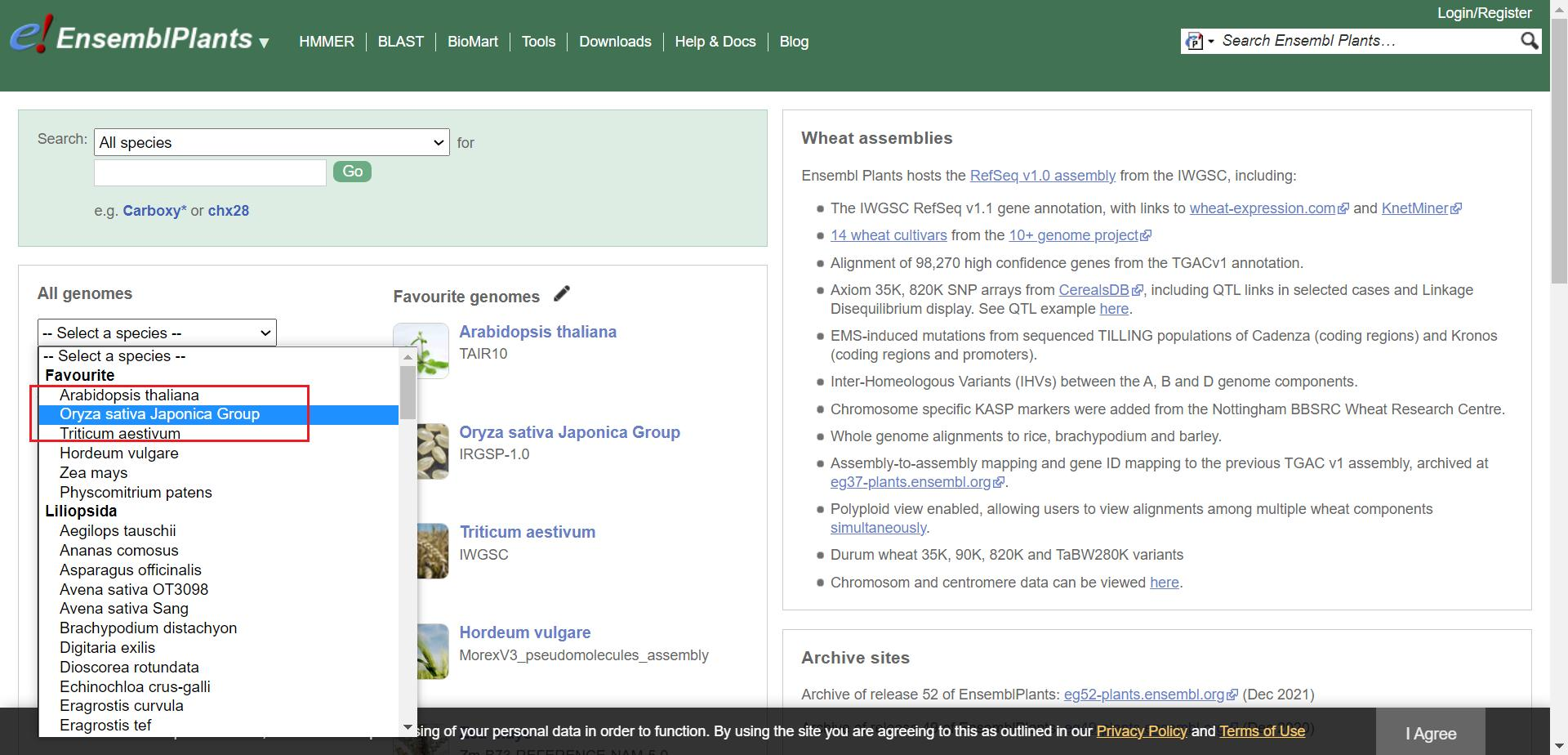

隐马尔可夫模型下载
隐马尔可夫模型 (Hidden Markov model, HMM) 是用于序列标注的概率图模型,描述一个隐藏的马尔科夫链生成不可观测的状态序列,再由每个状态生成一个观测而产生一个观测序列的过程,是一个生成模型。隐马尔可夫模型在自然语言处理、语音识别、模式识别等领域都应用广泛。在自然语言处理中,基于字标注的分词、词性标注、句法分析、命名实体识别等领域都可以应用隐马尔可夫模型。在生物学领域中,可以通过大量序列数据进行深度学习后生成隐马尔可夫模型,用于预测具有某个特征的基因家族。
简单来说,就是通过已知的同源基因序列,在算法中进行机器学习后生成一个隐马尔可夫模型,然后我们可以利用这个模型来预测其他物种中是否存在这样的同源基因序列。这样的隐马尔可夫模型其实就是 pfam 上的以 “.hmm” 为后缀的文件。我们在做基因家族鉴定的时候,用到的 hmmsearch,就必须要求输入这样的 hmm 文 件以及基因序列 fasta 文件。
还需要下载隐马尔科夫模型,进入 pfam 官网 ——keyword search:cystatin-GO——点击 PF00031——Curation&model——Download 下载 hmm 模型文件,此处下载的为 Cystain.hmm
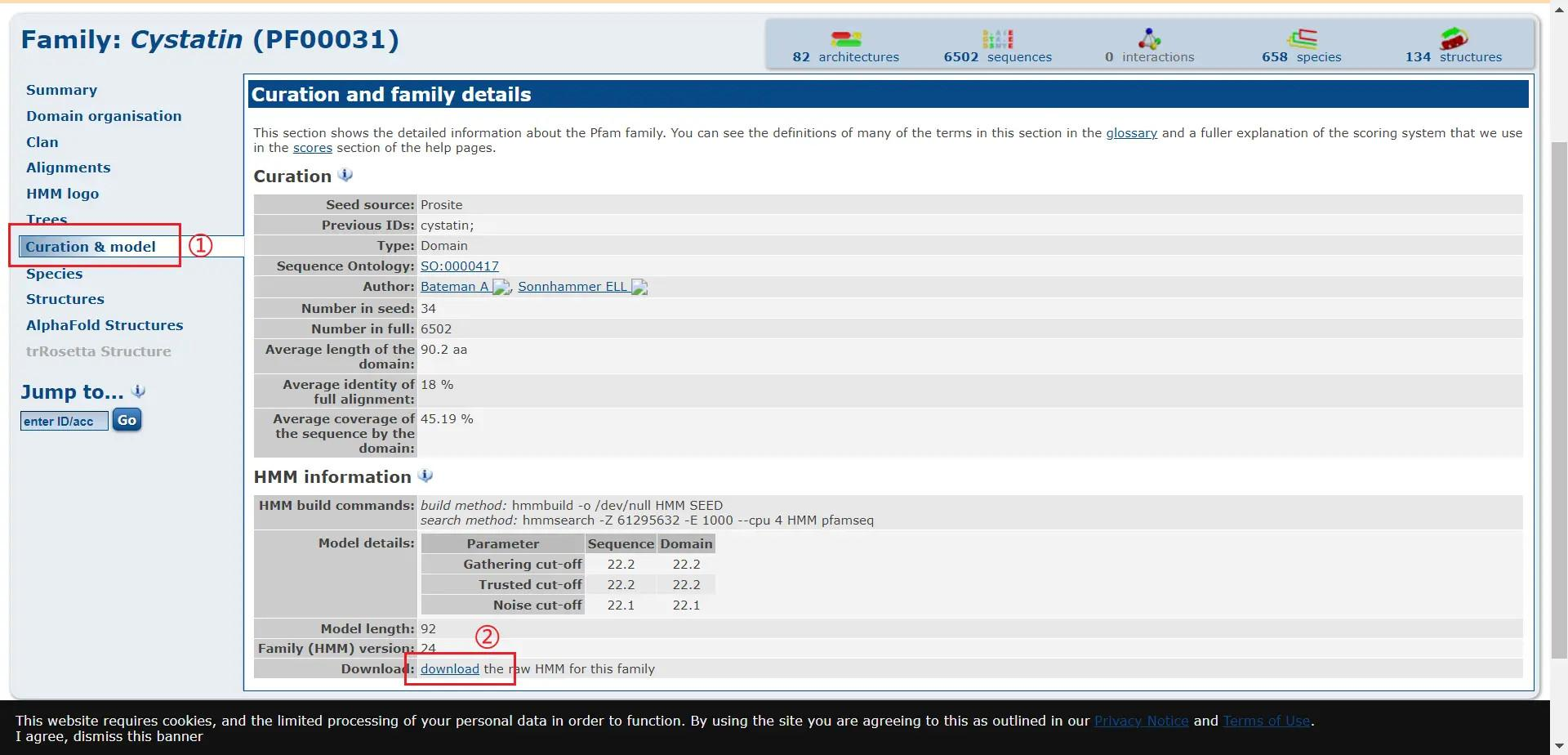
再点击 Alignments——Fomat 选择 stockholm-gengerate 下载 Stockholm file,此处下载的文件名为 PF00031_seed.txt

基因家族鉴定
我们要研究水稻 cystatin 基因家族,那么就需要先查找水稻中属于 cystatin 家族的基因然后对其进行鉴定和分析
HMMER 查找
可以直接使用我的项目进行自动化鉴定,可以自动化 hmmsearch 并且提取符合阈值的基因 ID,并且提取蛋白质,项目地址
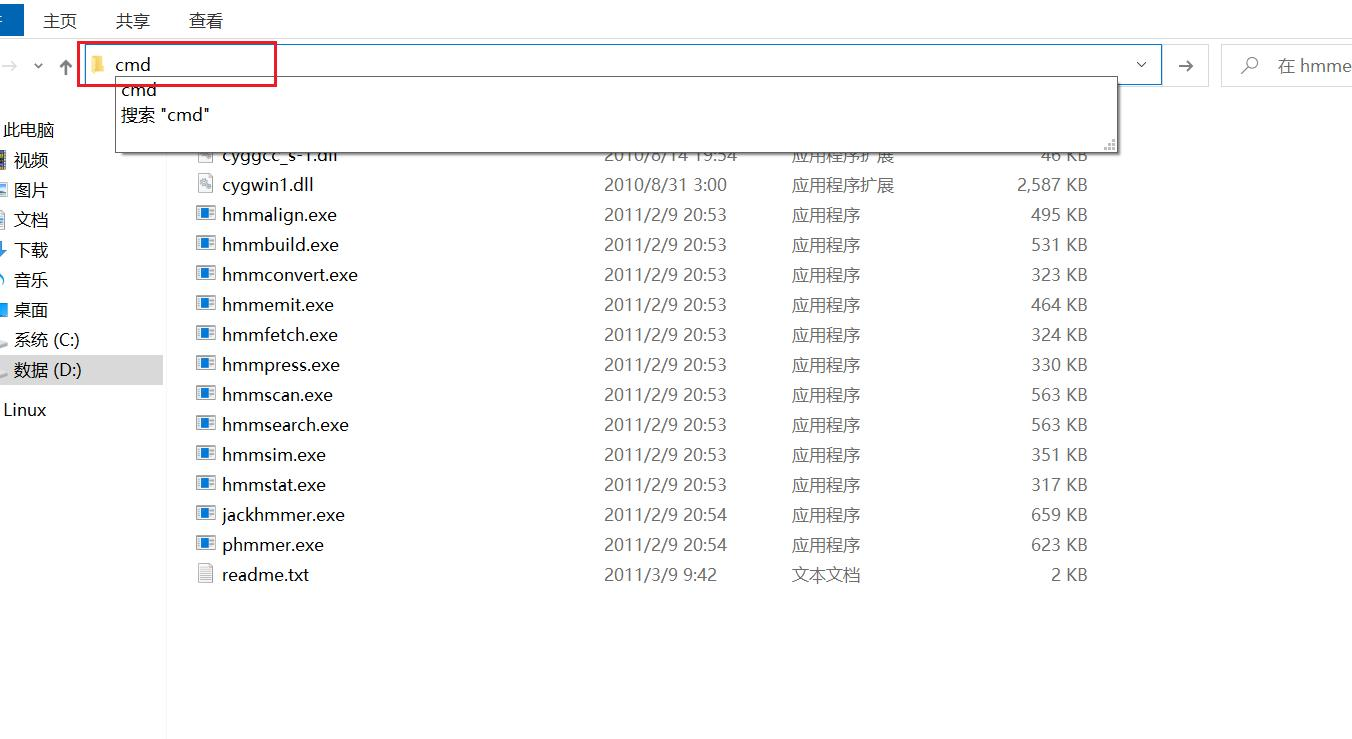
点击此链接下载 HMMER Windows 版本 hmmer-3.0-windows.zip 将压缩包解压在任意位置,进入 HMMER 文件夹,确保当前可以看到。exe 的可执行程序

在 Windows 资源管理器地址栏中输入 CMD 回车在此位置打开 CMD 命令行,命令与 Linux 系统下一致
构建参考数据的隐马尔可科夫模型
hmmbuild Cystain.hmm PF00031_seed.txt
基于隐马尔可科夫模型在整个水稻基因组的蛋白质序列中查找同源基因
hmmsearch Cystain.hmm allprotein.fasta > result.out
最终得到搜索结果如下图所示,我们可以通过这两列 E-value 来判断基因是否属于 Cystain 基因家族,下图中,最后三个基因的 E-value 均小于 10 的-5 次方,但们在某些情况下,我们可以将其先保留,进行进一步判断
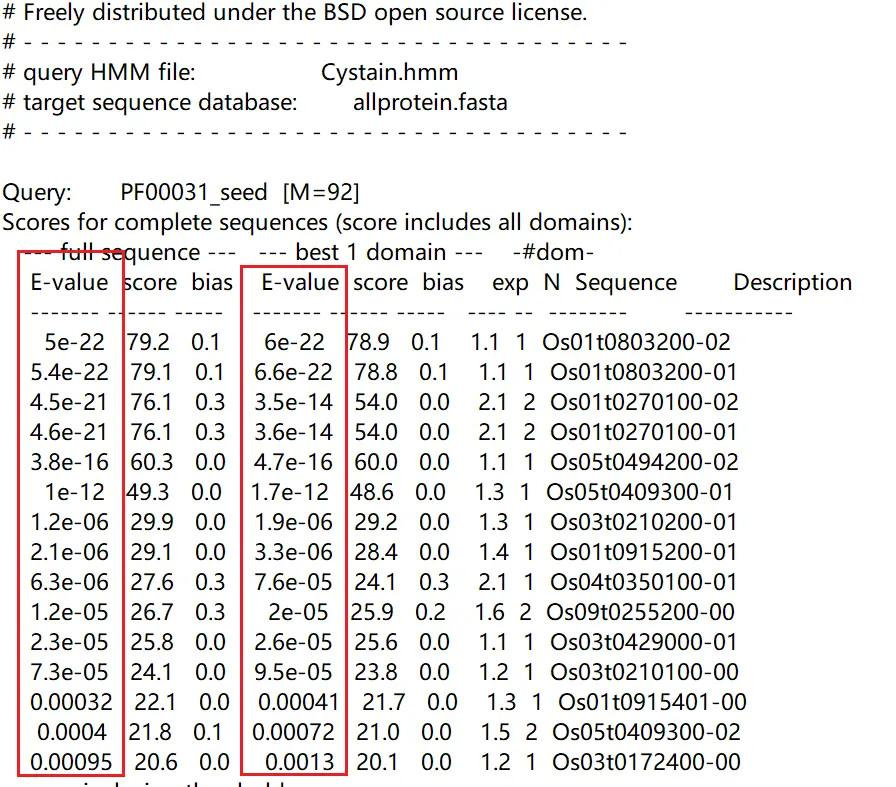
HMMER 的搜索结果显示水稻中 Cystain 基因家族有下面 14 个基因
| gene | gene |
|---|---|
| Os01g0803200 | Os01g0915200 |
| Os01g0270100 | Os05g0409300 |
| Os05g0494200 | Os04g0350100 |
| Os03g0429000 | Os03g0210200 |
| Os03g0172700 | Os03g0210100 |
| Os09g0255200 | Os02g0278600 |
| Os03g0210000 | Os01g0915401 |
Blast 比对
我们现在要对 HMMER 搜索到的基因进行 blast 比对,确保 HMMER 搜索结果是准确的
首先我们要获取这 14 个基因的蛋白质序列,使用 RAP-DB 网站获取或者 TBtools 翻译 CDS 序列都可以
然后使用 TBtools 提取基因组的全部 CDS 序列,再使用 TBtools 的 Batch Translate CDS to Protein 功能将 CDS 序列翻译为蛋白质序列,作为序列比对的参考序列
打开 TBtools 的 Blast Compare Two Seqs 工具,输入查询序列、基因组序列并设置输出文件,将输出格式改为 Table 方便查看,然后进行比对
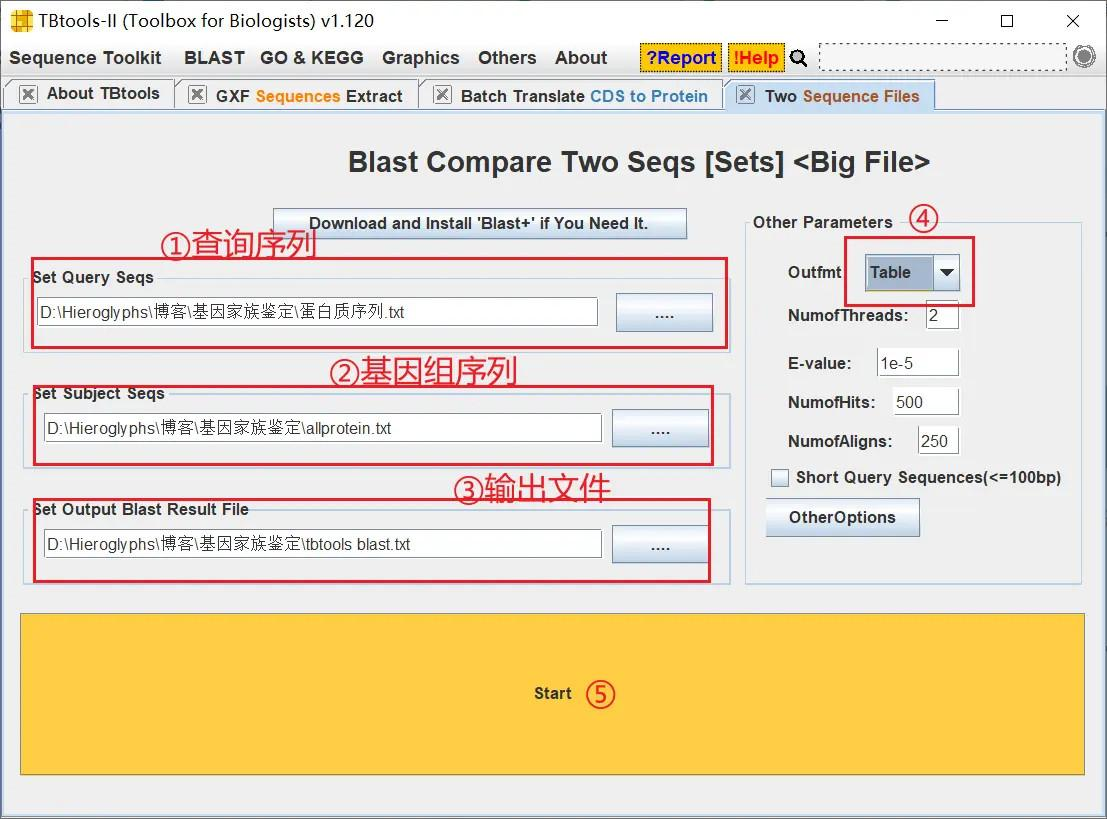
得出结果后,查看 evalue 这一列,所有的值均小于 10 的-5 次方,我们现在可以认为 HMMER 搜索结果是准确的,这 14 个基因都属于 Cystain 基因家族
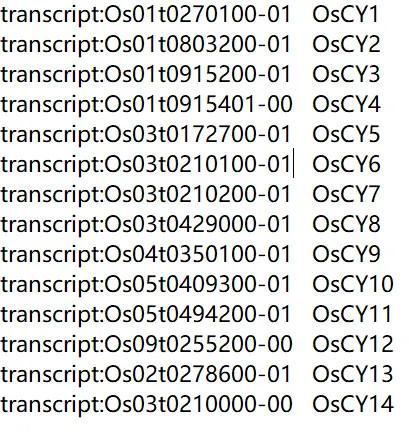
 这里我对以上已经基本确定的 14 个基因进行重命名,如下表,名称参考于文献 《拟南芥和水稻 cystatin 基因家族的生物信息学分析_杨泽峰》 中的命名方式
这里我对以上已经基本确定的 14 个基因进行重命名,如下表,名称参考于文献 《拟南芥和水稻 cystatin 基因家族的生物信息学分析_杨泽峰》 中的命名方式
| Os ID | Rename | Os ID | Rename |
|---|---|---|---|
| Os01g0270100 | OsCY1 | Os03g0429000 | OsCY8 |
| Os01g0803200 | OsCY2 | Os04g0350100 | OsCY9 |
| Os01g0915200 | OsCY3 | Os05g0409300 | OsCY10 |
| Os01g0915401 | OsCY4 | Os05g0494200 | OsCY11 |
| Os03g0172700 | OsCY5 | Os09g0255200 | OsCY12 |
| Os03g0210100 | OsCY6 | Os02g0278600 | OsCY13 |
| Os03g0210200 | OsCY7 | Os03g0210000 | OsCY14 |
进一步确定基因家族成员
结构域分析
先使用 NCBI CD Search 进行结构域的查找

然后使用 TBtools 进行可视化
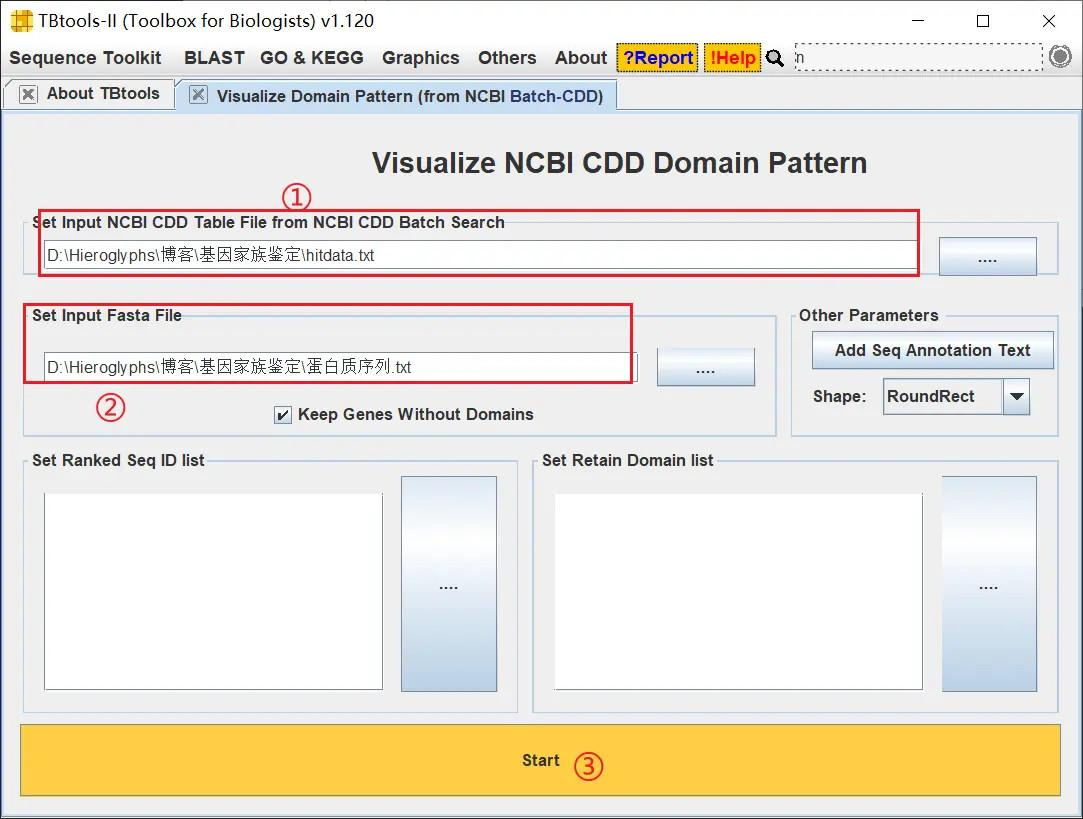
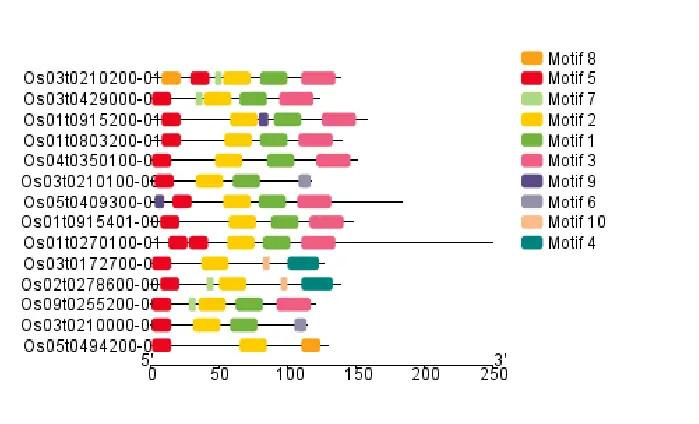
通过 TBtools 绘制的图像可以看出,这些基因是全部具有 Cystatin 结构域的
Motif 分析
将候选基因的蛋白质上传至 MEME 网站

然后使用 TBtools 进行可视化
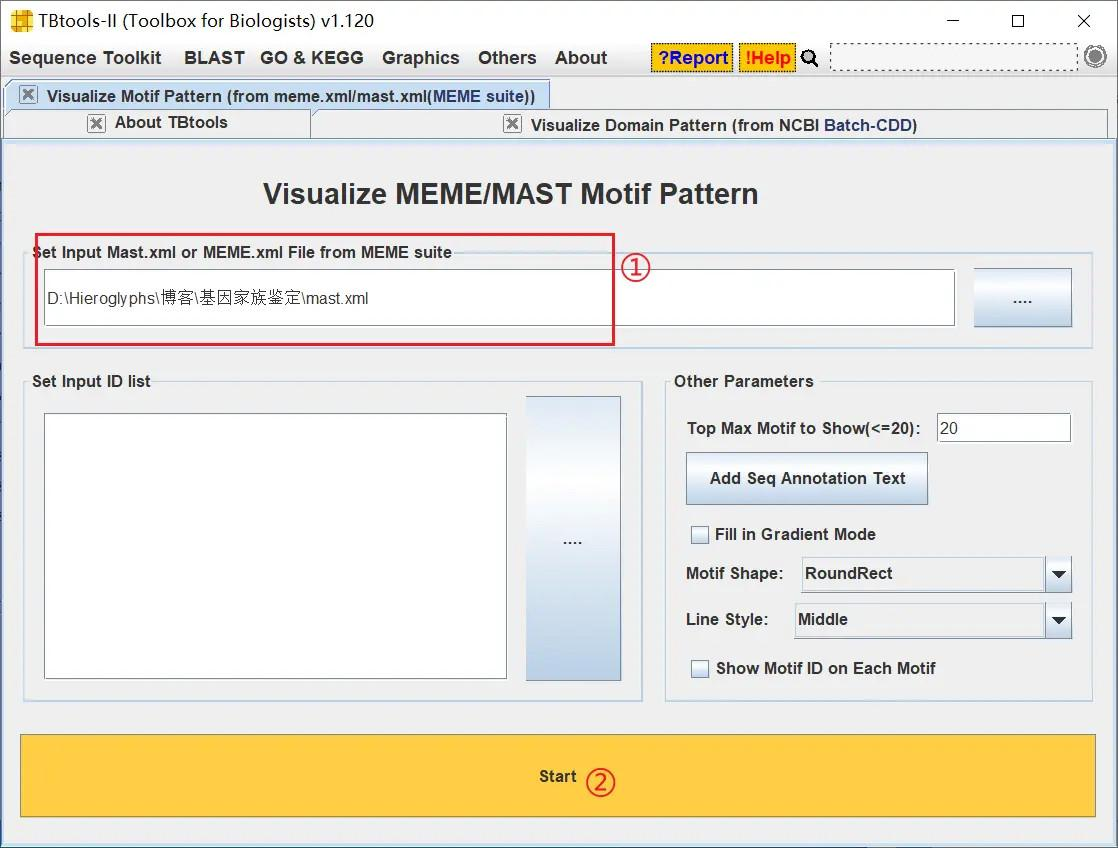
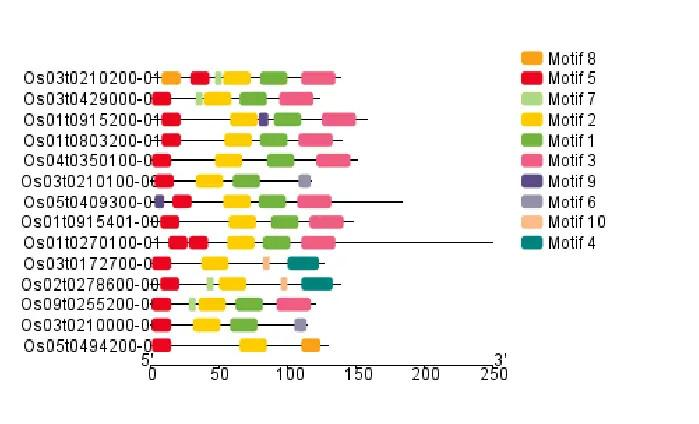
从 TBtools 绘制的图像可以看出,这些基因的保守基序大致上也是比较相近的
刚刚用 TBtools 做的 domain 分析和 motif 分析的图像是不太清楚、不怎么美观的,后面我们可以通过 TBtools 中多图结合的功能来将它们做的更加美观
构建系统发育树
构建系统发育树需要使用 mega7 软件,打开此页面,填写相关信息、选择版本后下载
https://www.megasoftware.net/older_versions
将所有选定基因的蛋白质序列放在一个文件中,将拓展名改为 .fasta ,双击使用 mega7 打开
按 Ctrl+A 全选,点击 Alignment,选择使用 ClustalW 算法进行多序列比对
比对完成后点击左上角的 Data,选择 Phylogenetic Analysis 进行进化树分析

进化树分析完成后,点击上方 Phylogeny,选择 NJ 算法构建进化树

这个进化树貌似并不是很美观,我们为保证图的美观性和统一性,要把进化树中基因名字根据前文所命名的样式进行重命名,直接在比对所使用的 fasta 文件中进行修改,将 > 后的名称改为我们重命名的名称即可
然后点击 File 里面的 export current tree(Newick),点开后点 export,再点击保存图标保存进化树的结果,后面我们可以使用 evoliview 或者 iTOL 网站对保存的结果进行美化
从其系统发育树来看,只有 Os05t0594299-01 这一个基因的发育与其它基因相差较大
由以上步骤我们可以确定,候选基因全部属于 Cystatin 基因家族,接下来就要对其性质进行分析
基因家族的性质分析
理化性质分析
打开 TBtools 的 Protein Paramter Calc 功能,导入蛋白质序列文件,并设置输出文件,点击 Compute 进行分析
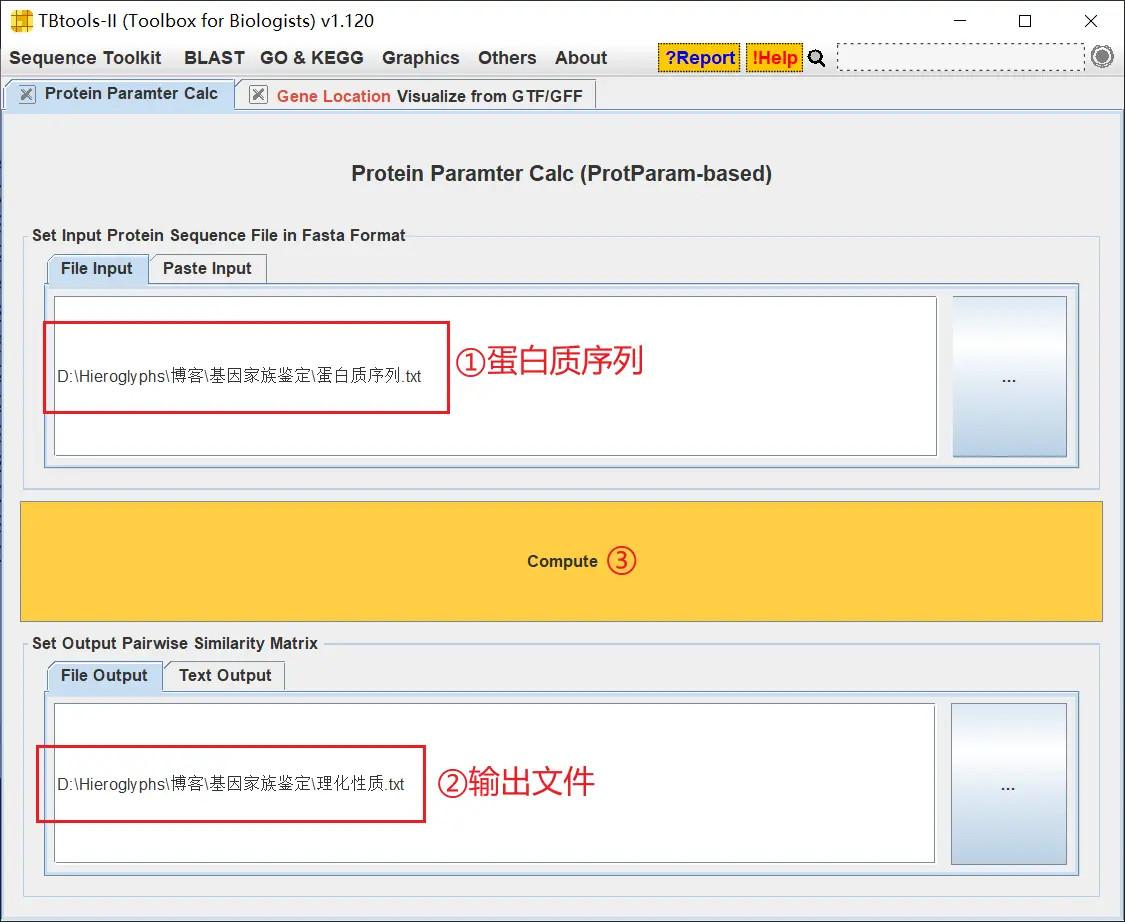
得到分析结果

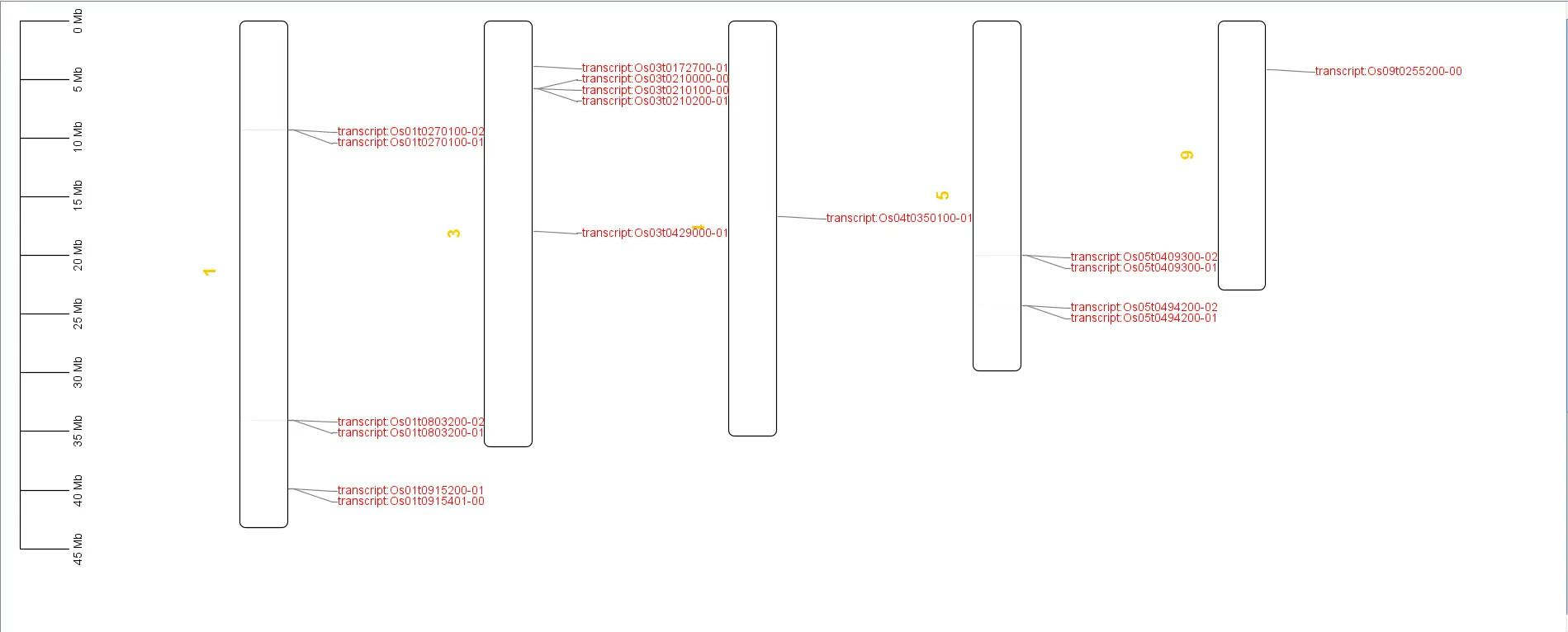
染色体定位分析

得到染色体位置分布图
我们导入重命名文件,使其名称全部变为我们命名的格式
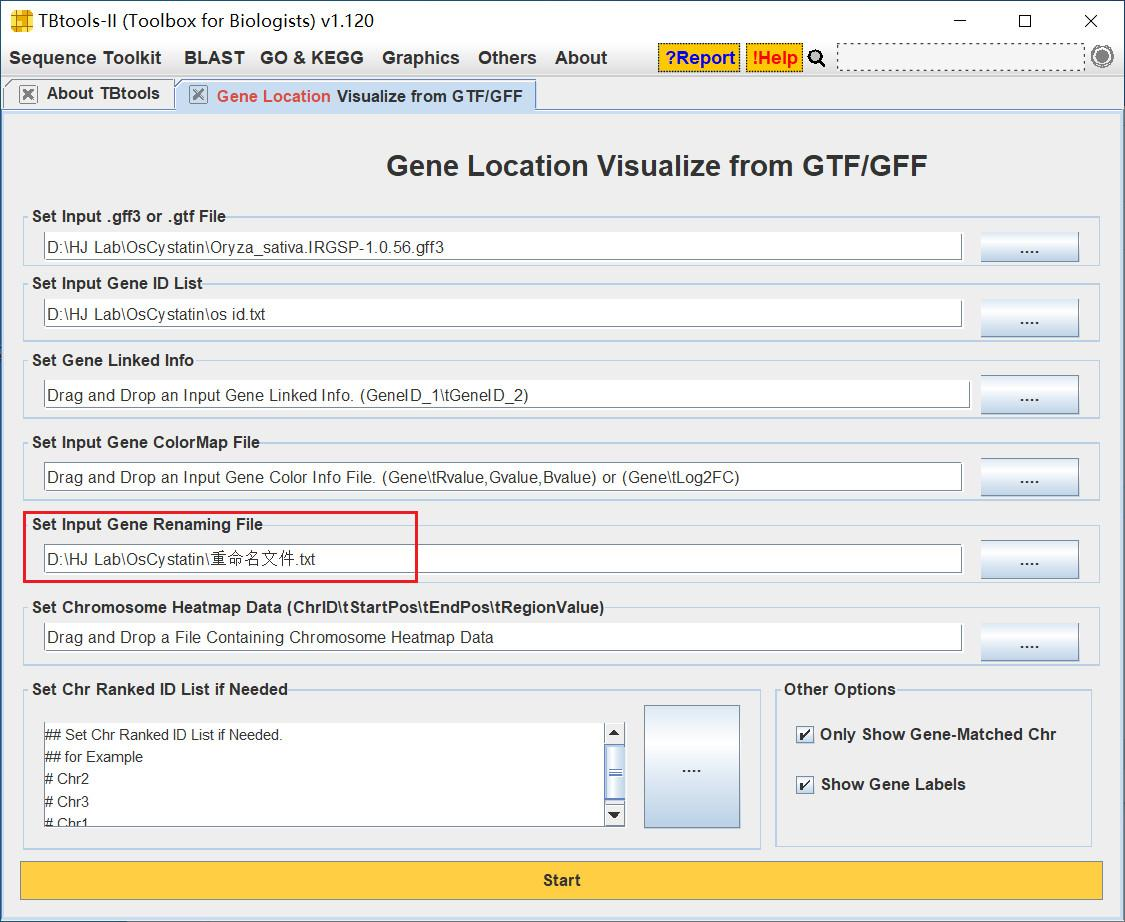
注意重命名文件的格式
可以在这里对颜色和字体进行简单的美化
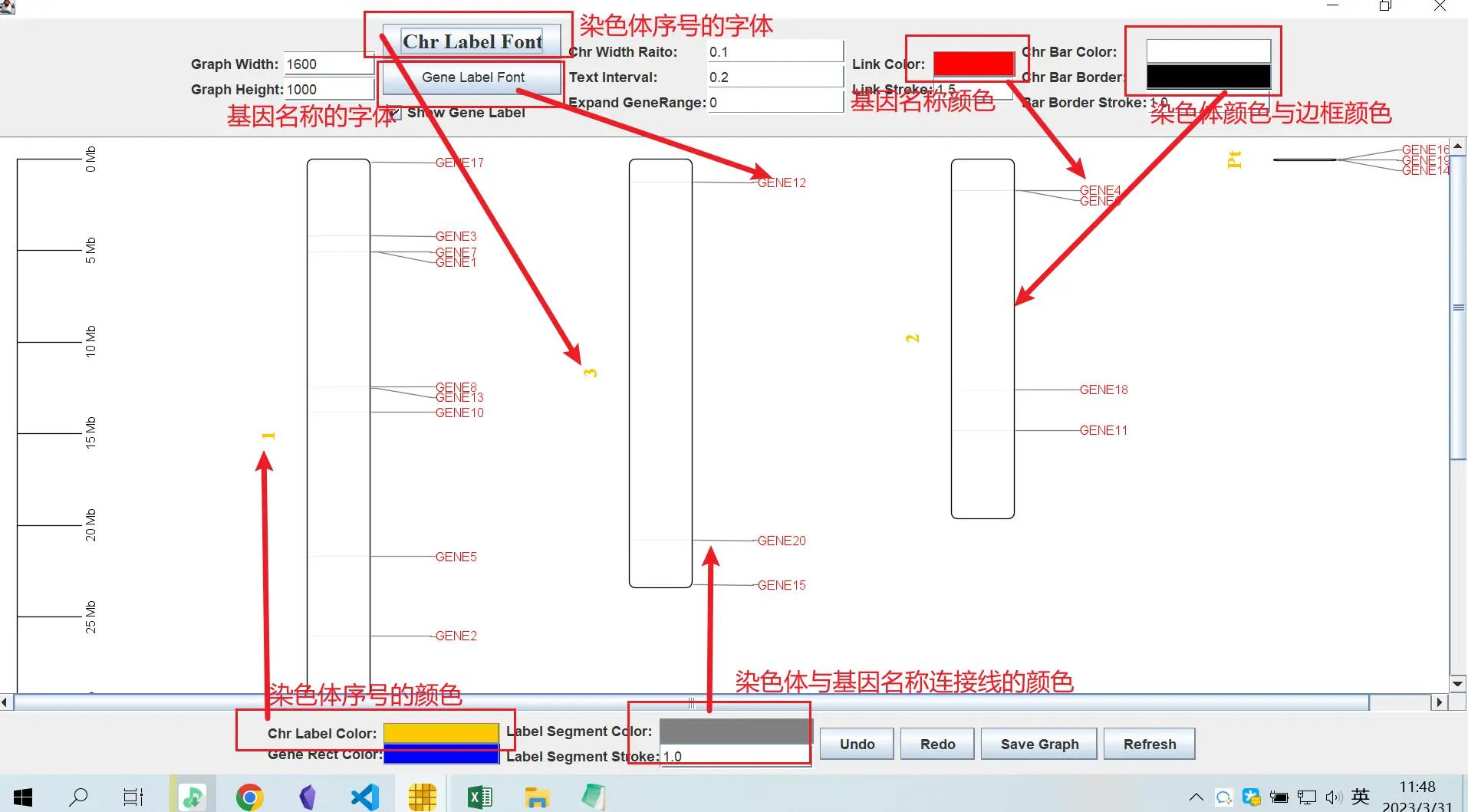
当然,如果你不想对它们进行统一的更改,可以点击任意一个基因名称,再点击右键对其进行单独的更改,对于染色体的名称也同样适用
调整过后的图就好看了很多

基因家族成员的基因分析
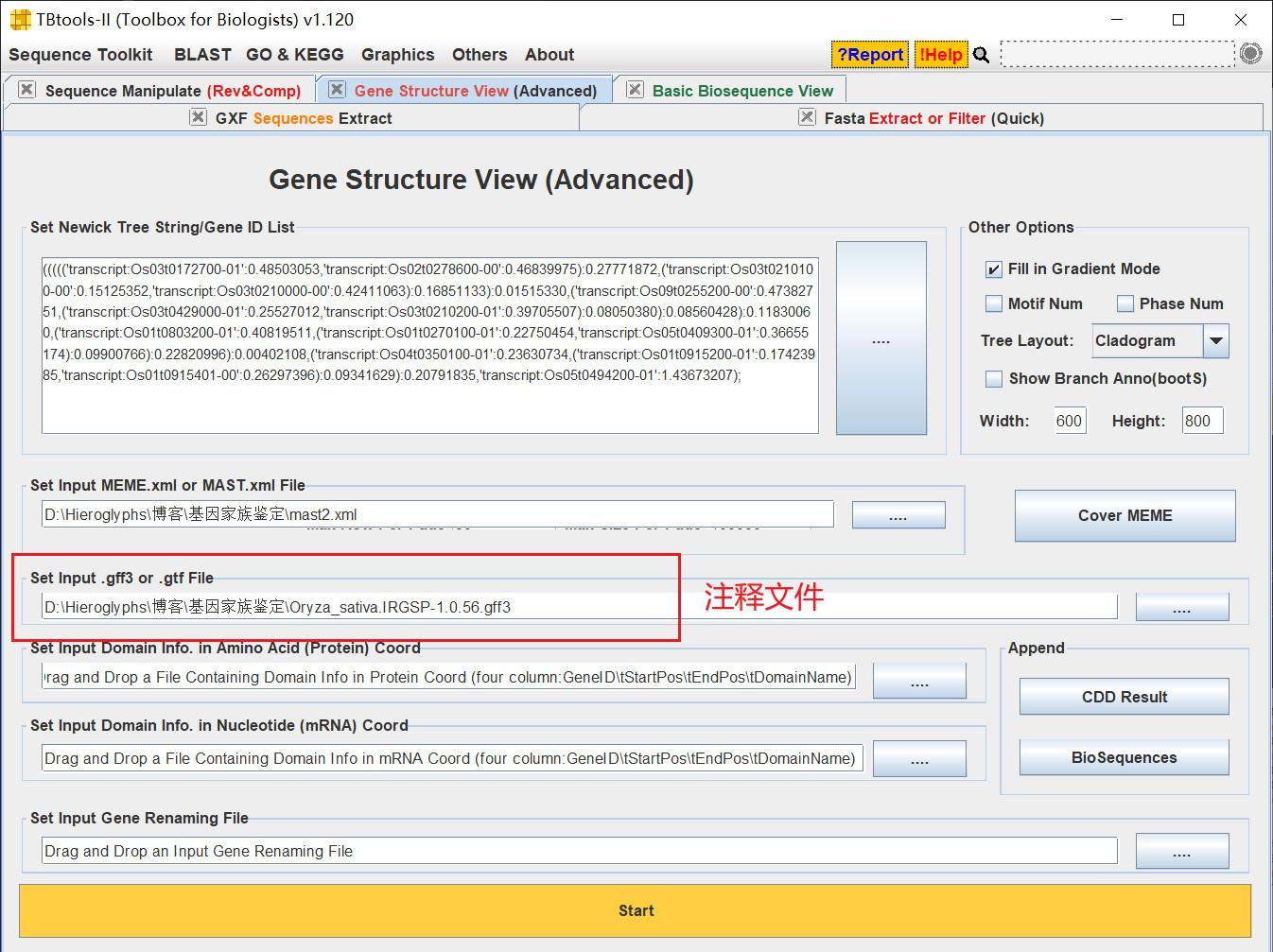
内含子和外显子结构
这里使用的也是 TBtools 中多图结合的插件,打开 Gene Structure View 功能,粘贴基因列表,导入注释文件后即可得到内含子和外显子的位置信息

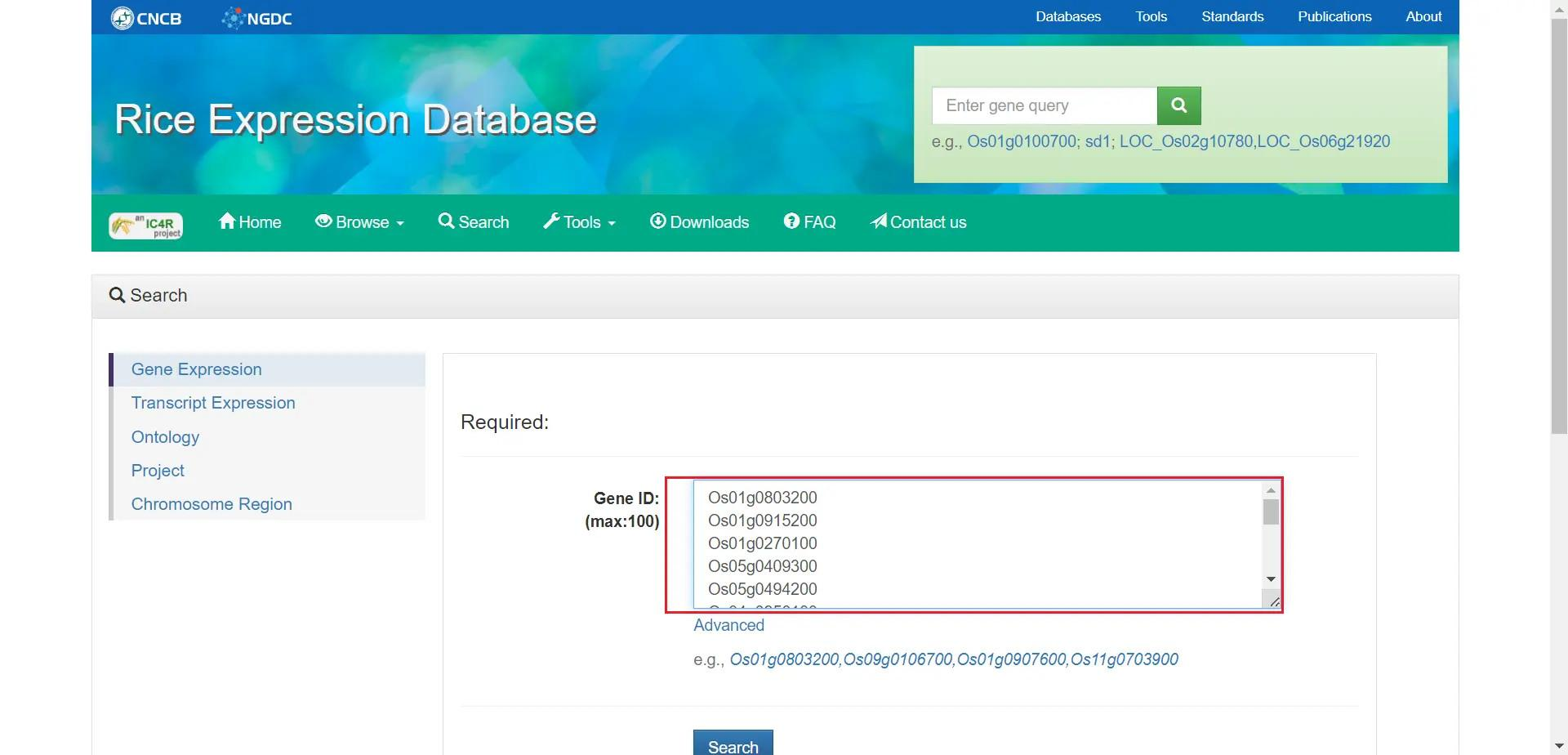
基因表达量分析
首先在 Rice Expression Database 中搜索基因列表
选择 DRP000391 这个项目
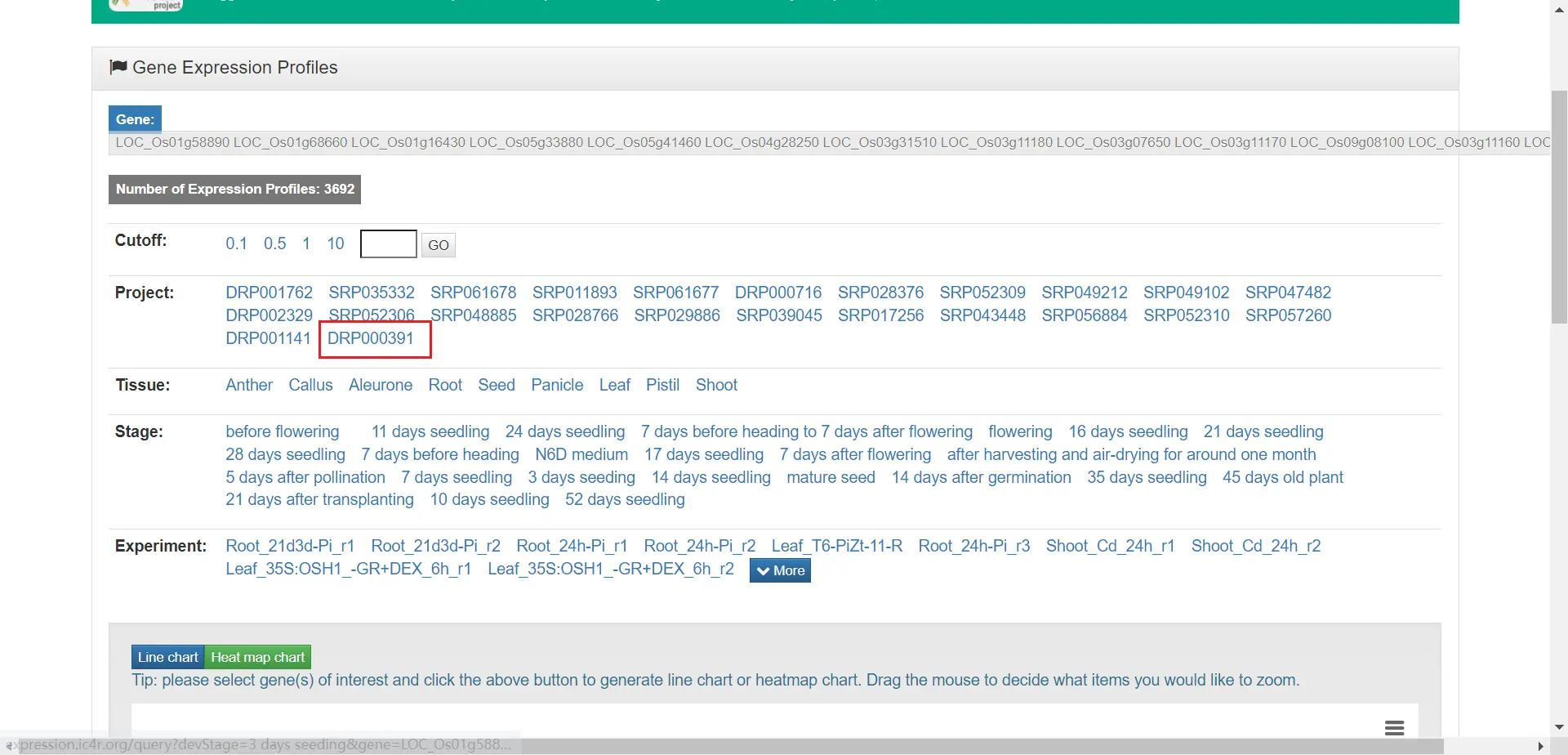
随后点击 Show data —— Export —— CSV 导出数据,由于 Chrome 和 Edge 早已不支持使用 Flash,所以这里用 360 极速浏览器导出
用 Excel 打开数据,在 Tissue 这一列进行筛选,分别获得基因在水稻的 Callus、Leaf、Panicle、Panicle、Root、Seed、Shoot 等部位的表达数据,如果有些数据有两个,那么求平均值
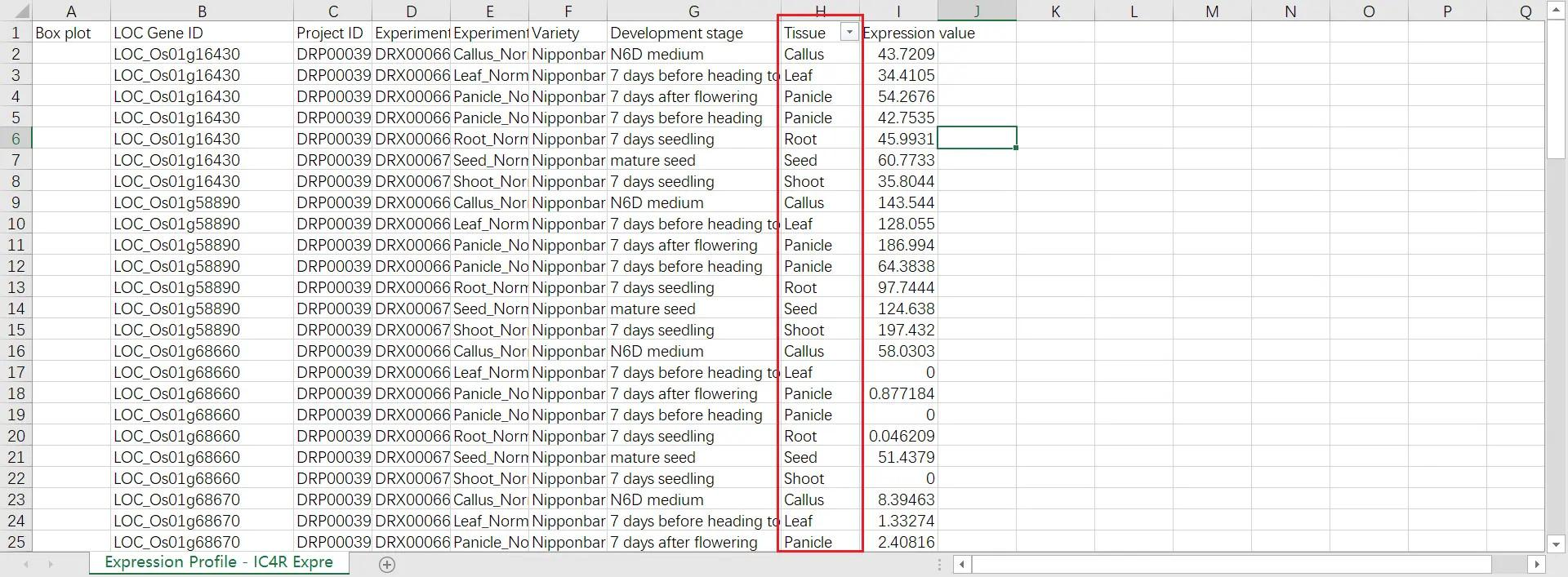
整理成如下样式

然后对数据进行 LOG2 处理,如果出现#NUM! 字样,全部改为 0
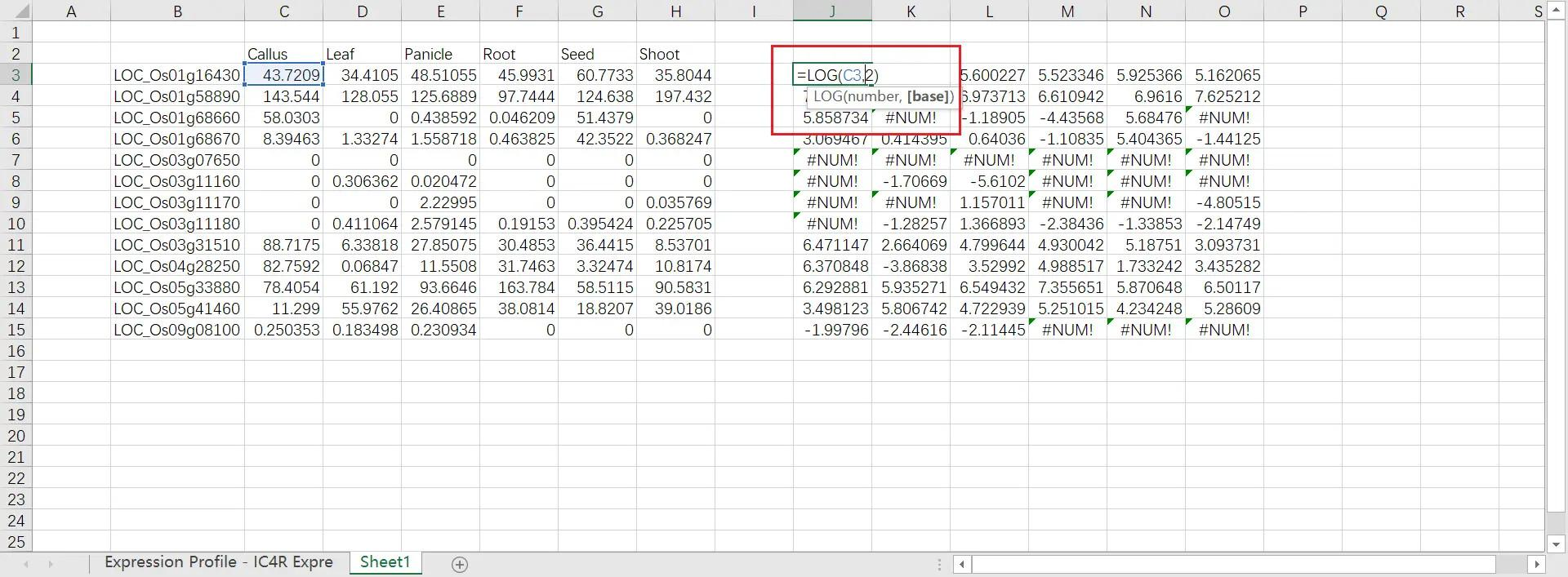
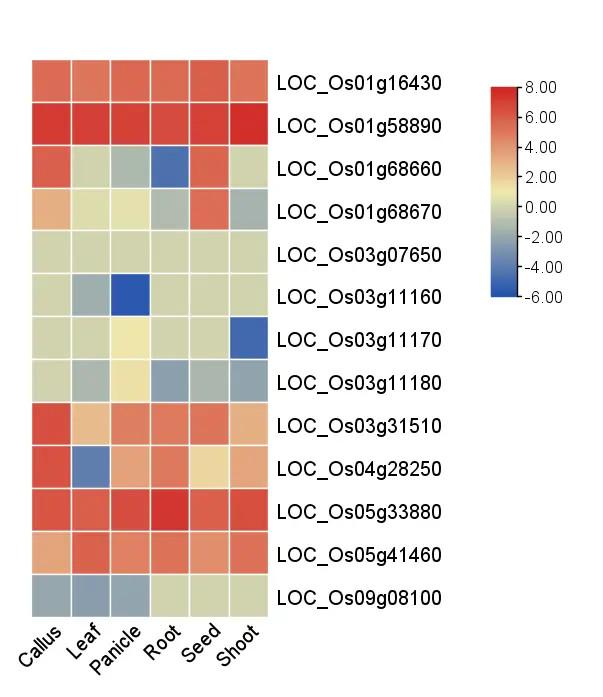
重新整理得到最终数据,打开 TBtools 的 Heat Map 功能,把刚刚整理的数据粘贴进去进行绘图

得到基因家族的表达热图
对基因进行重命名即将复制进去的表格中的基因名称进行修改即可
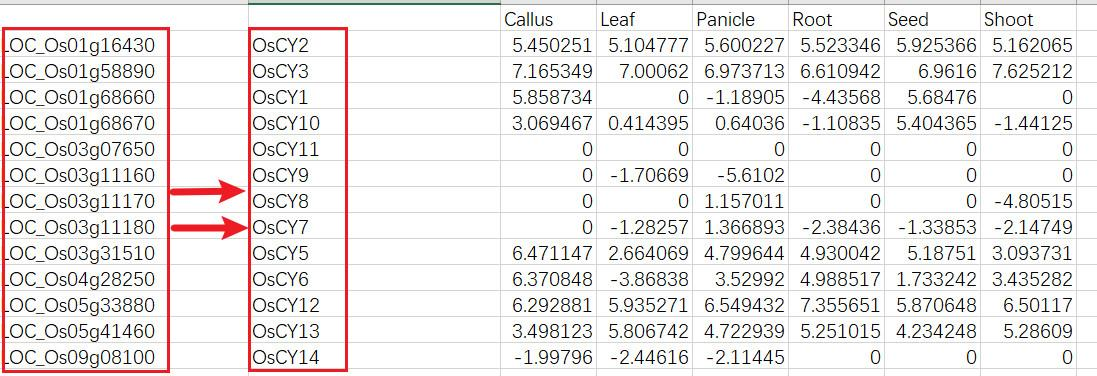
然后重新制图
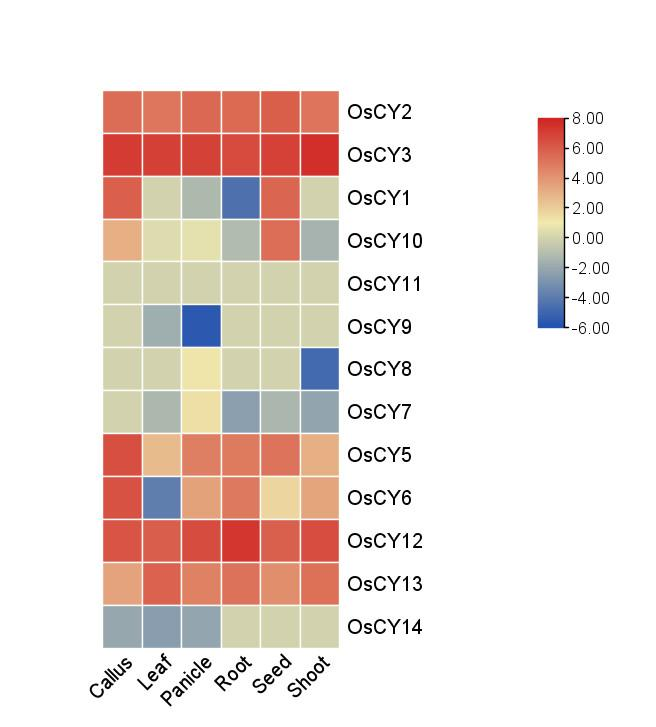
也可以对图像进行美化
基因家族的进化分析
互作蛋白网络
打开 string 数据库 ,点击 search ,输入蛋白质的 Os ID 并选择蛋白质的物种,点击 search

搜索完成后就得到了蛋白质的互相作用网络
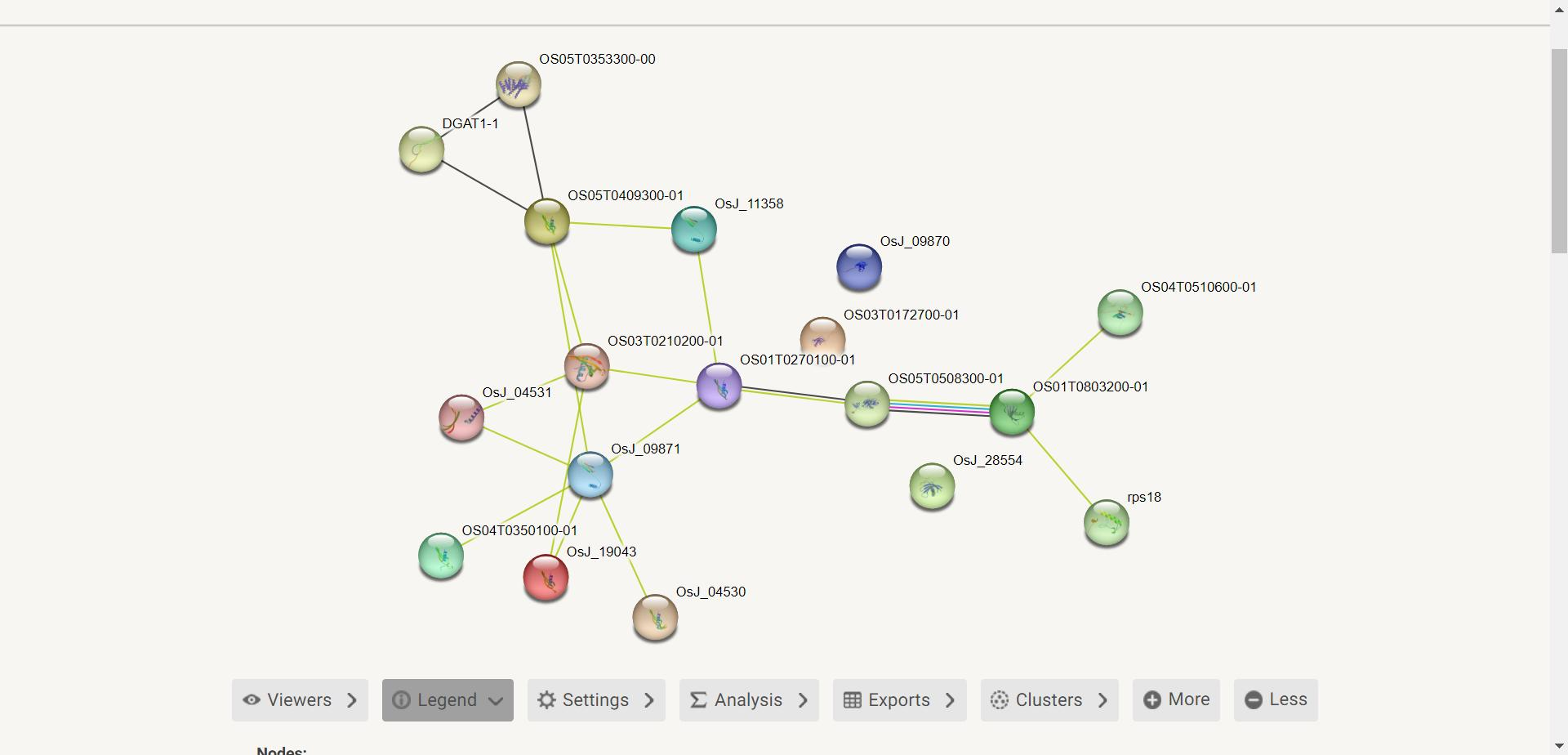
点击每一个圆珠我们就可以查看其信息,包括基因 ID、物种等,圆珠直接的黑色线条表示共表达蛋白质,黄色线条表示在其他生物体中共同提及推定的同源物。

顺式作用元件分析
顺式作用元件 (cis-acting element) 存在于基因旁侧序列中能影响基因表达的序列
顺式作用元件包括启动子、增强子、调控序列和可诱导元件等,它们的作用是参与基因表达的调控。顺式作用元件本身不编码任何蛋白质,仅仅提供一个作用位点,要与反式作用因子相互作用而起作用
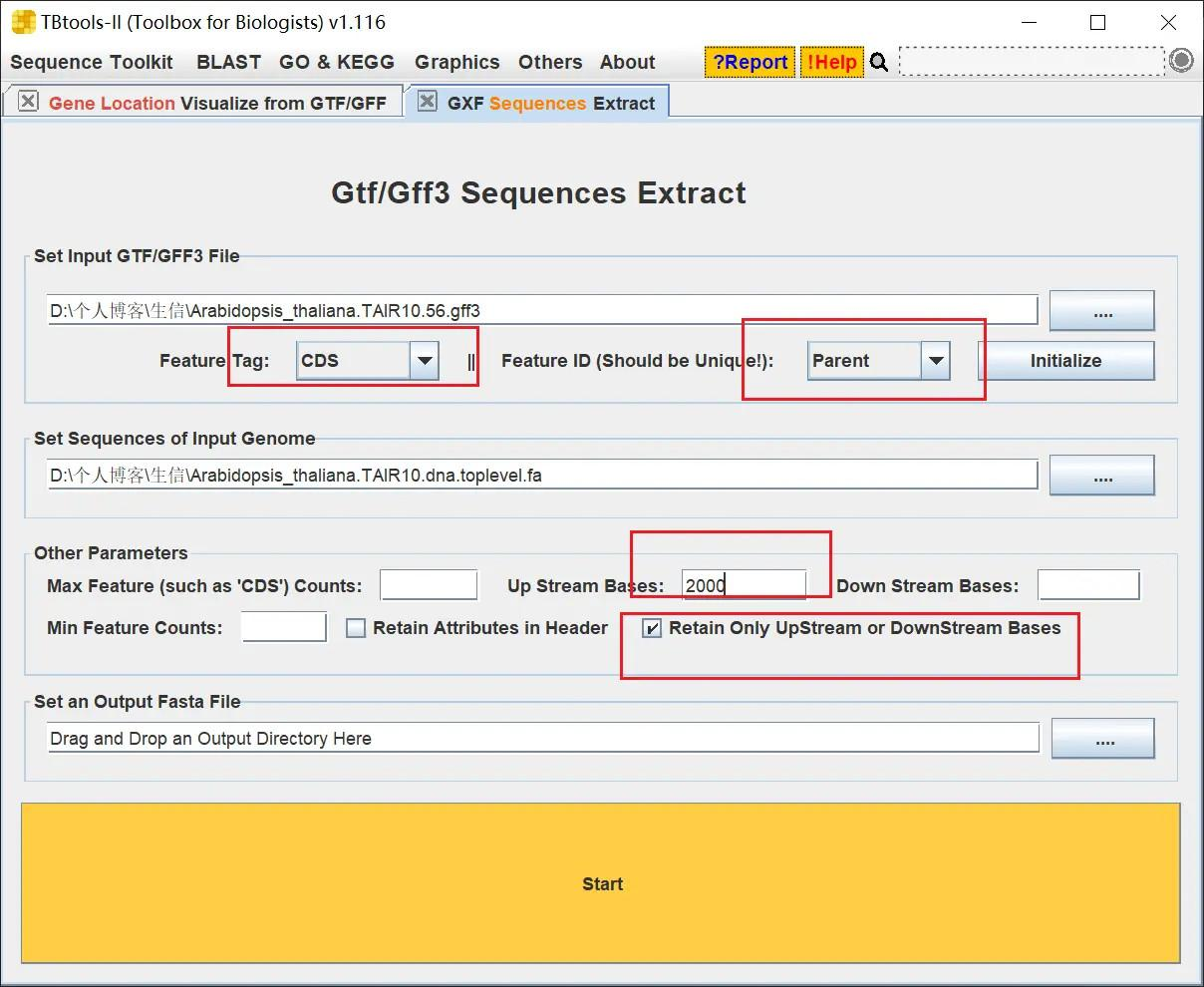
首先要做的第一步是提取所有基因的 CDS 序列
提取时需要在初始化后将 TBtools 的设置改为如下配置
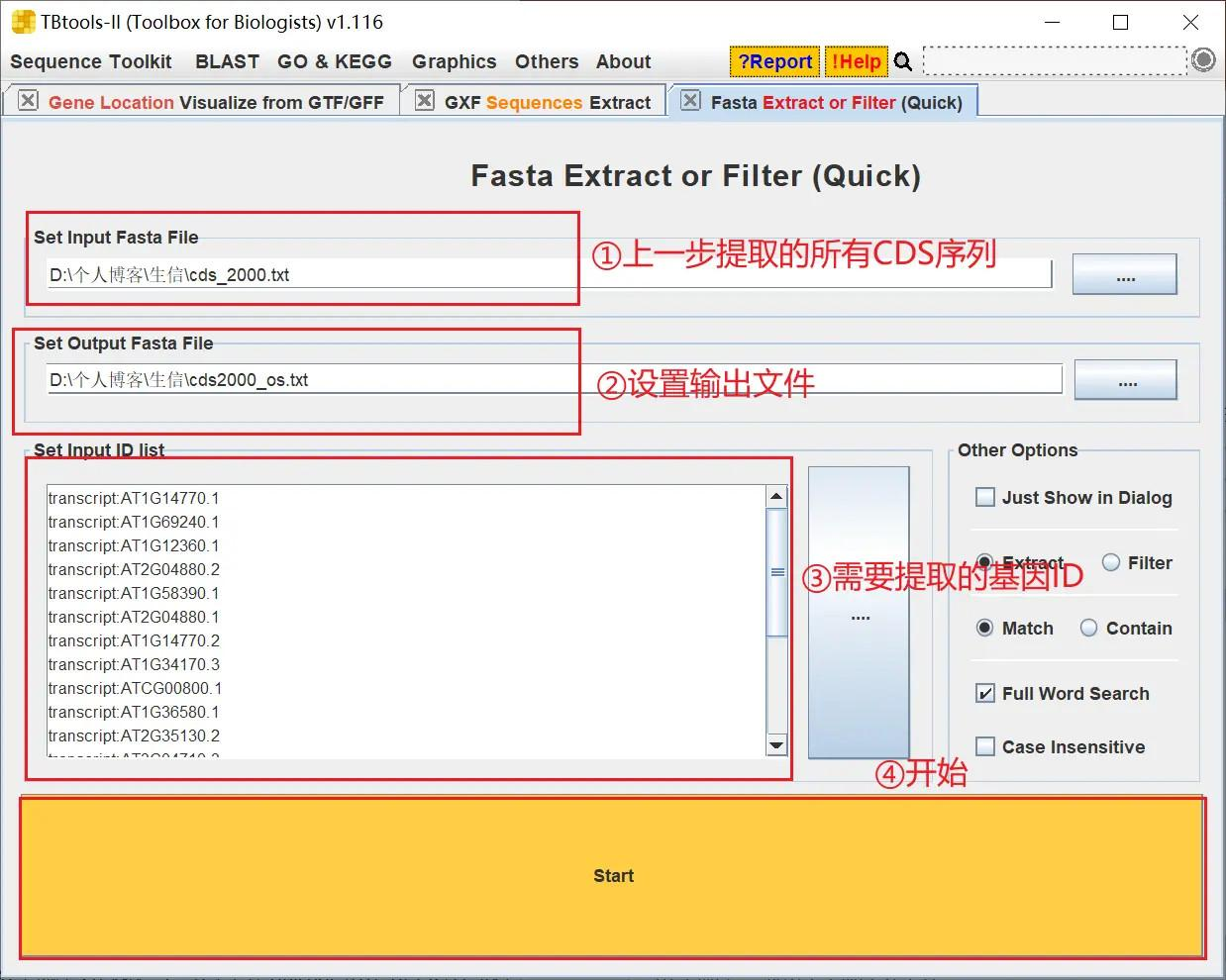
然后提取目标基因的启动子序列,打开 TBtools 的 Fasta Extract or Filter,设置好序列文件、输出文件、基因 ID,点击 start 开始
打开 Sequenxe Manipulate 工具,将提取出来的所有序列复制进去,全部转为大写

新建文本文档保存转换后的序列
现在需要打开网站 http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ 将刚刚保存的序列上传进行顺势作用元件预测


等待一会后会收到网站发出的一封邮件,下载邮件中的附件然后解压,解压之后得到一个拓展名为 tar 的文件,再进行解压
用 excel 打开解压出来的 tab 文件,此时 excel 中显示的就是这些基因的元件信息
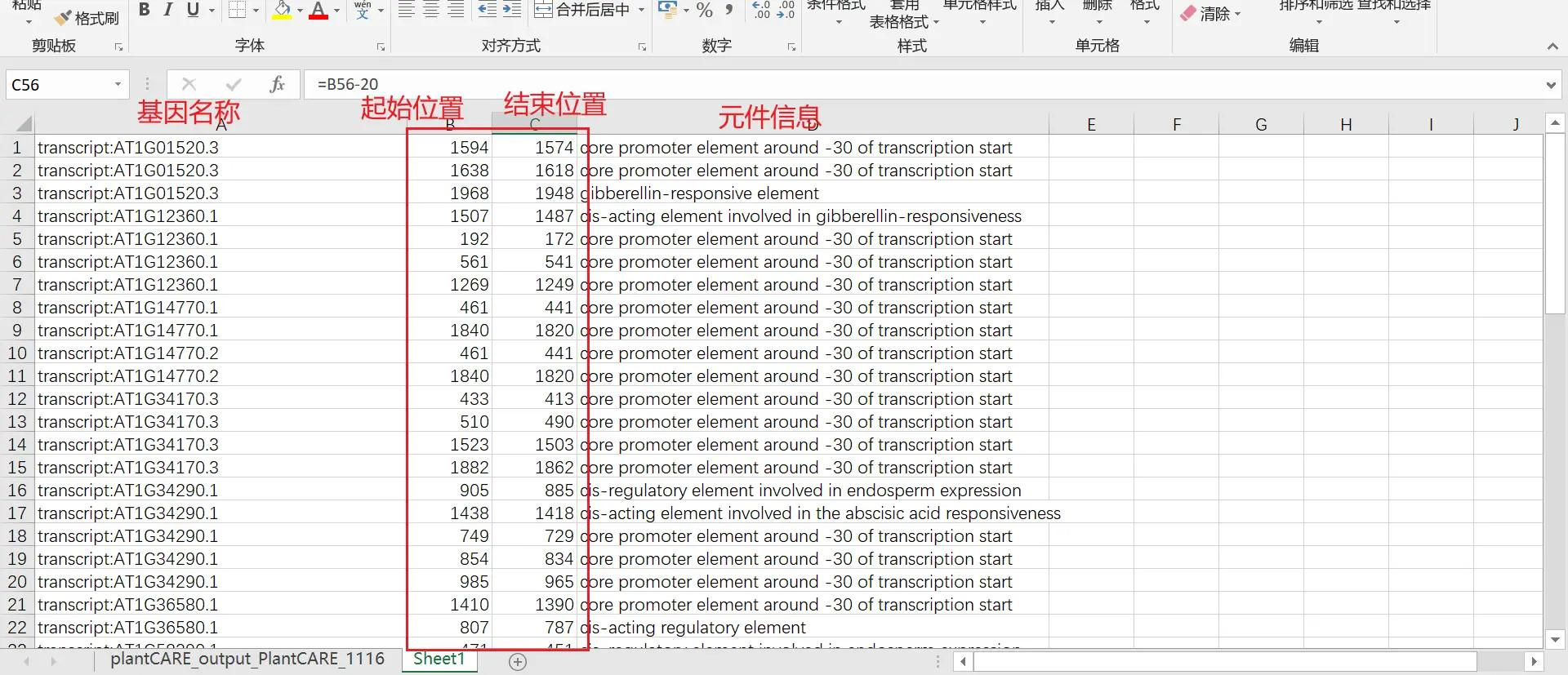
筛选我们需要的物种,这里我选择了水稻
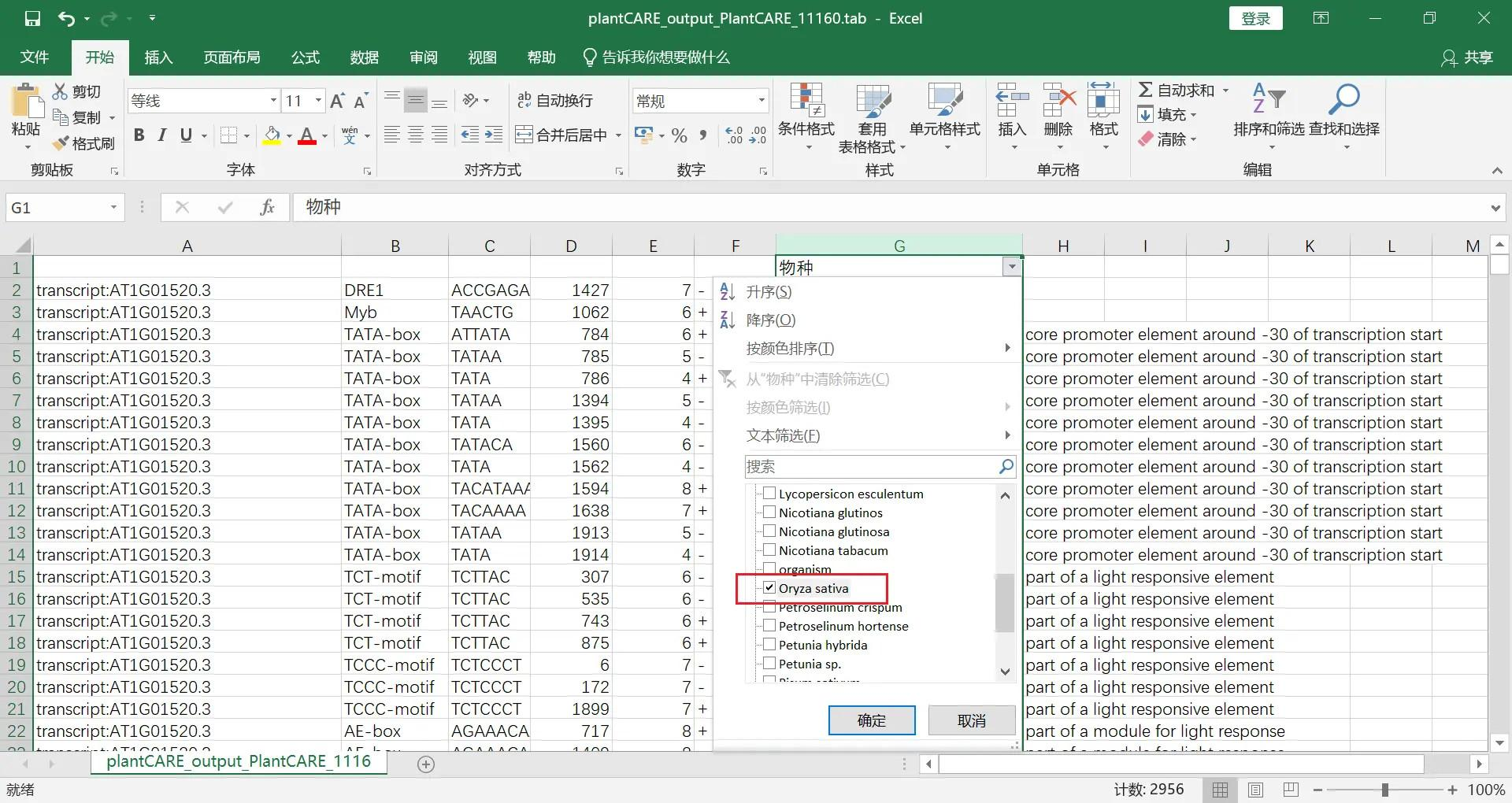
删除元件名称、元件序列、增减个数、加/减、物种等我们不需要的信息,然后为了保证最后的图片可看,这里需要扩大一下起始位置,较大的可以减少 20,较小的可以增大 20。并且需要将含有空白信息的项删除,不然 TBtools 会报错。最终整理成如下格式:

下面需要再新建一个关于序列长度的文件,格式如下:
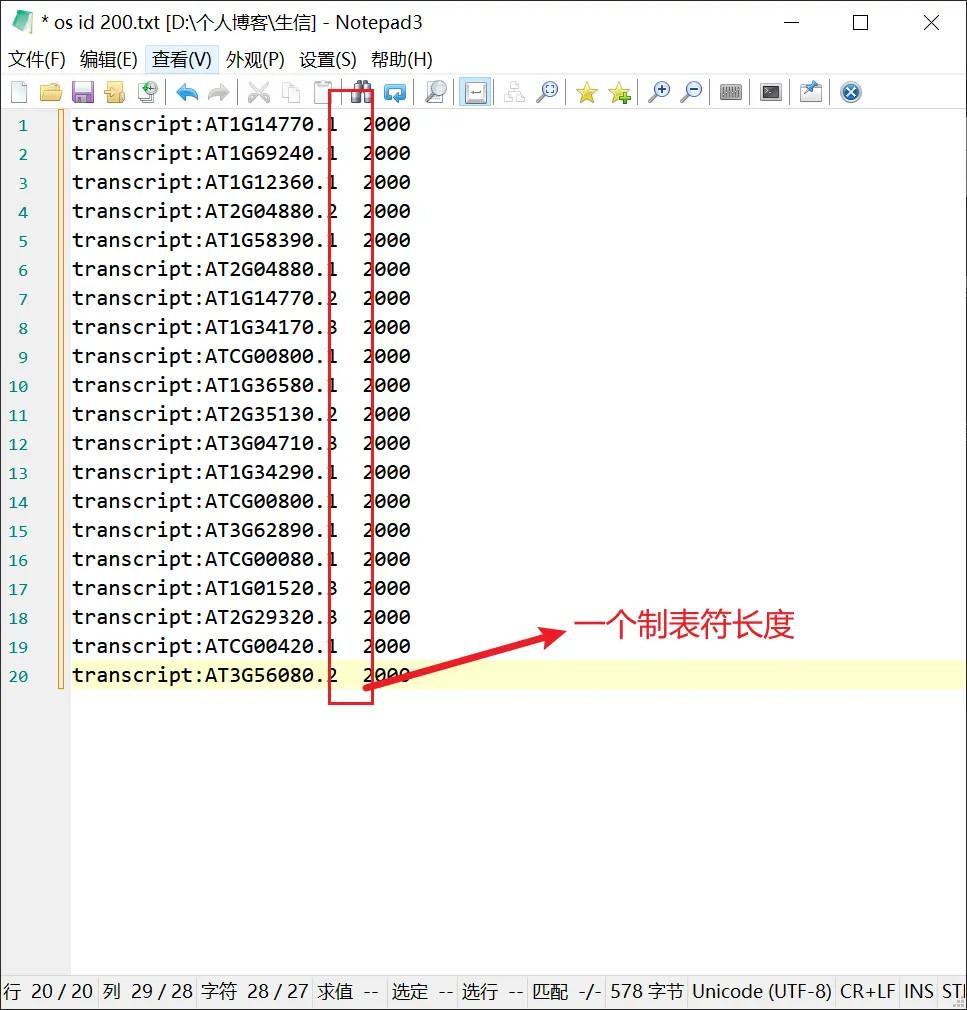
打开 TBtools 的 Simple BioSequence Viewer 功能,导入长度信息文件和元件信息,点击 start 开始
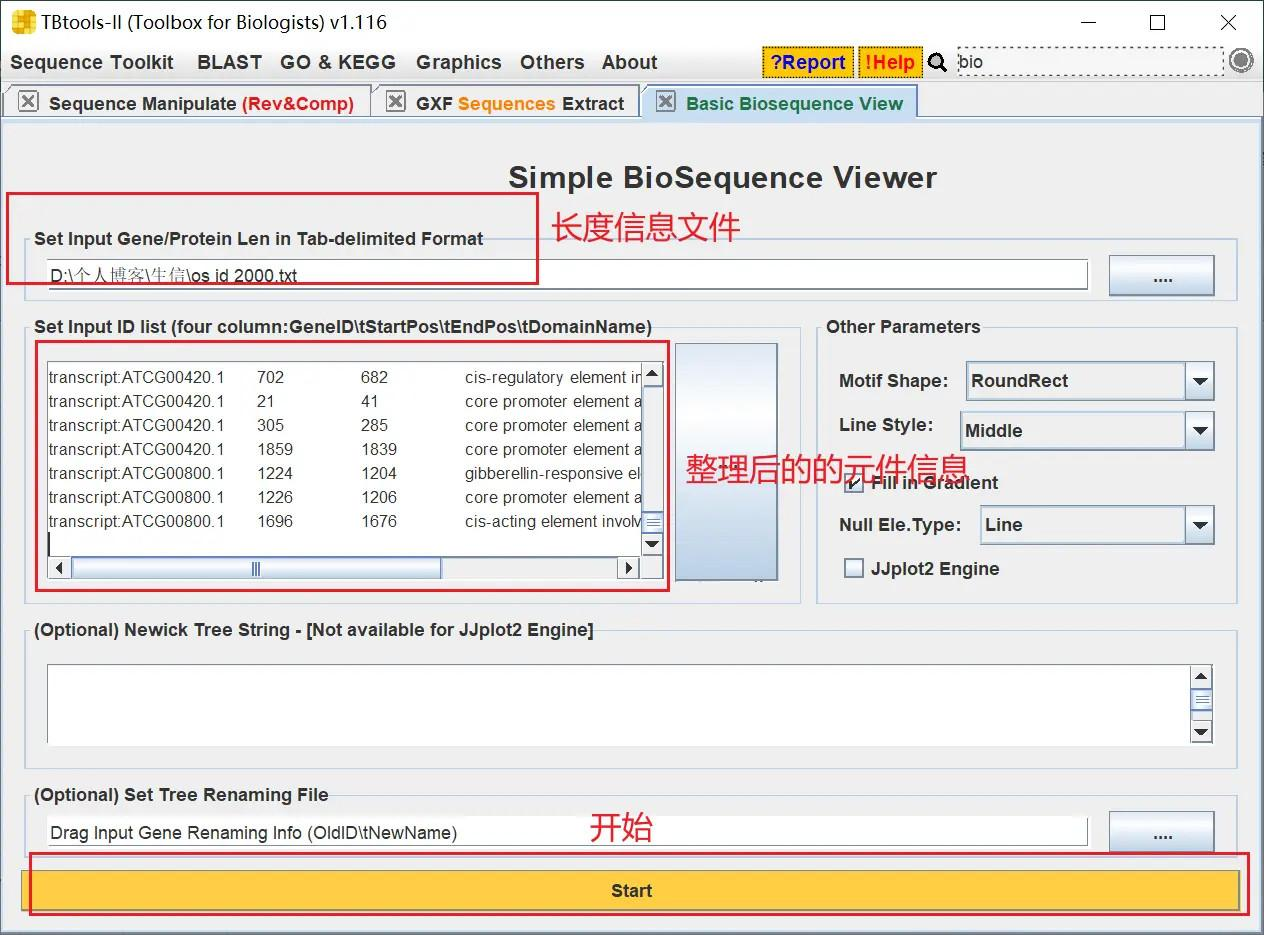
最终得到可视化结果
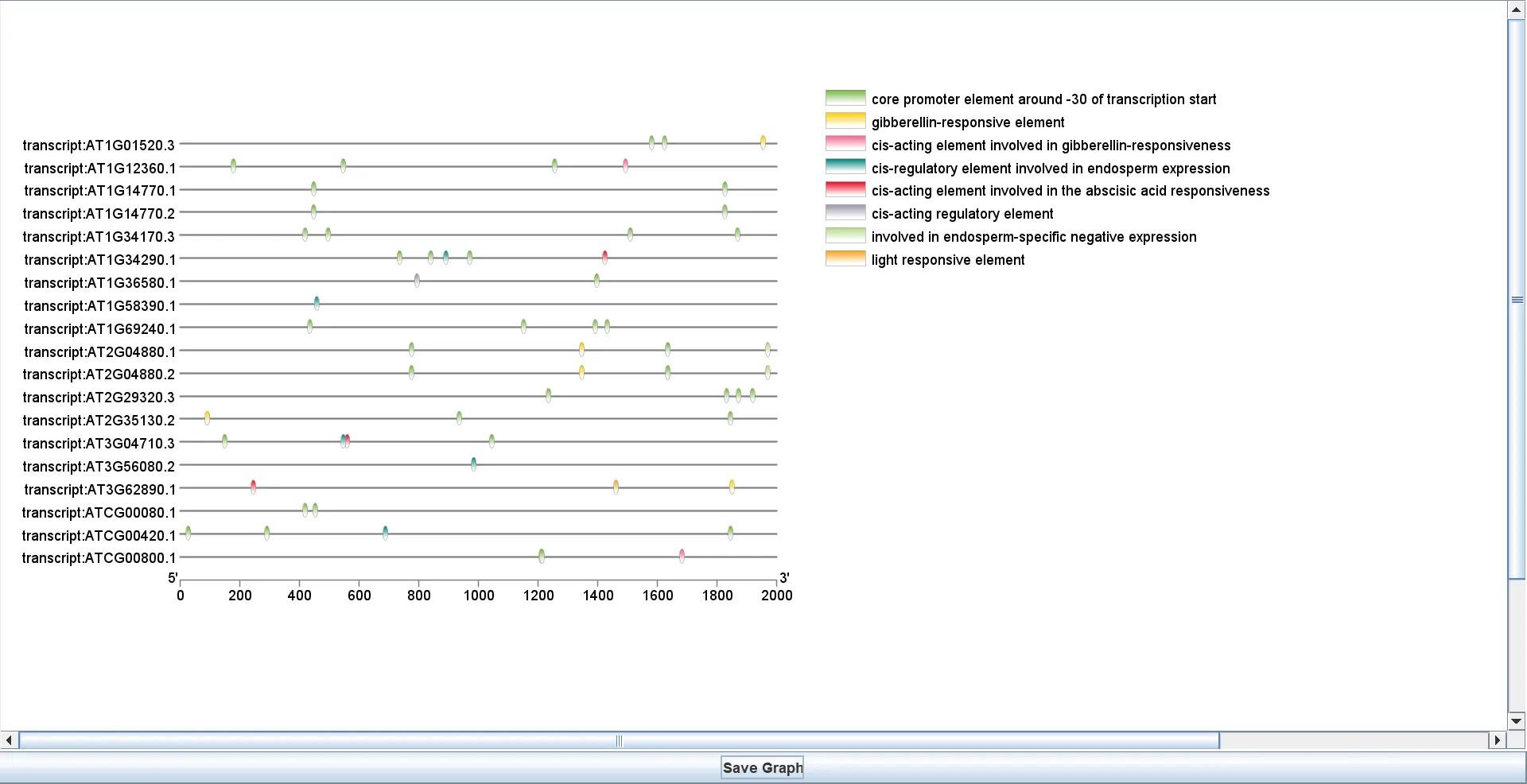
这是我做的顺式作用元件分析的图,可以对其进行简一些美化

多序列比对
在 CLUSTALW 网站 上传基因家族的蛋白质序列进行多序列比对,比对完成后下载比对结果的aln 文件

打开 ESPript ,上传刚刚的aln 文件进行可视化
下载结果中的 pdf 文件打开后即可得到多序列比对的图像

与进化树相似,修改多序列比对中的基因名称直接在比对的蛋白质序列中进行修改即可
共线性分析
共线性是指同源基因在物种内或者物种之间的分布或排列关系。我们都知道,有相当多的基因在一个物种中不是以单拷贝的形式出现,而是多拷贝,具有基因序列重复事件,也就是说能行使相同功能的同源基因在同一个物种内可能不只有一条序列,而当我们想研究这些同源基因在同一个物种内的重复事件的时候,就是物种内的共线性分析。如果我们研究同源基因在不同物种内的分布情况,就是物种间的共线性分析,物种间可以是两个物种,也可以是多个物种。
共线性分析可以用 MCScanX 软件进行分析,但是这个软件的官网已经 404 了,而且利用命令行来进行分析比较麻烦,这里建议大家使用 TBtools 上的工具,这个软件里面集成了共线性分析的相关插件,并可以进行可视化操作,分析起来很方便。
共线性分析最基本的原理是基于对基因序列进行 blast 比对,然后利用 MCScanX 软件对比对结果进行分析,这时候就需要有物种基因组的注释文件和比对结果,注释文件是用来分析这些基因所在的位置和数量,最后用可视化的软件把共线性分析的结果以图片形式展示出来。
TBtools 软件、物种基因组序列、注释文件
打开 TBtools 的 Fasta Stats 功能,导入物种基因组序列文件,设置好输出文件,获取染色体长度文件
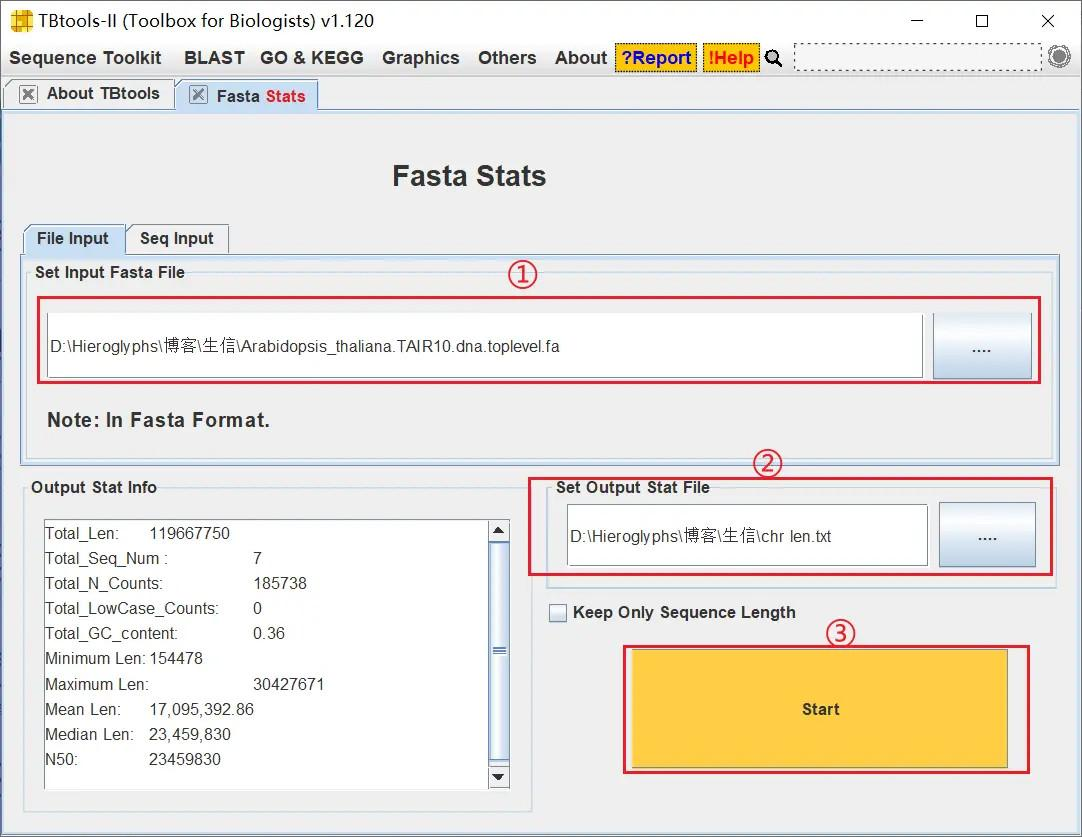
将 chr len.txt 文件的内容粘贴到 Excel,只保留 ID 和 length 两列的数据,删除 chr len.txt 文件中原有的数据,将 ID 和 length 两列数据保存在 chr len.txt 中
打开 TBtools 的 One Step MCScanX 功能,这里它需要输入两个物种的基因组序列文件和注释文件,我们两个都导入拟南芥的进行物种内的共线性分析,这个过程中如果出现报错,关掉即可,不用理会

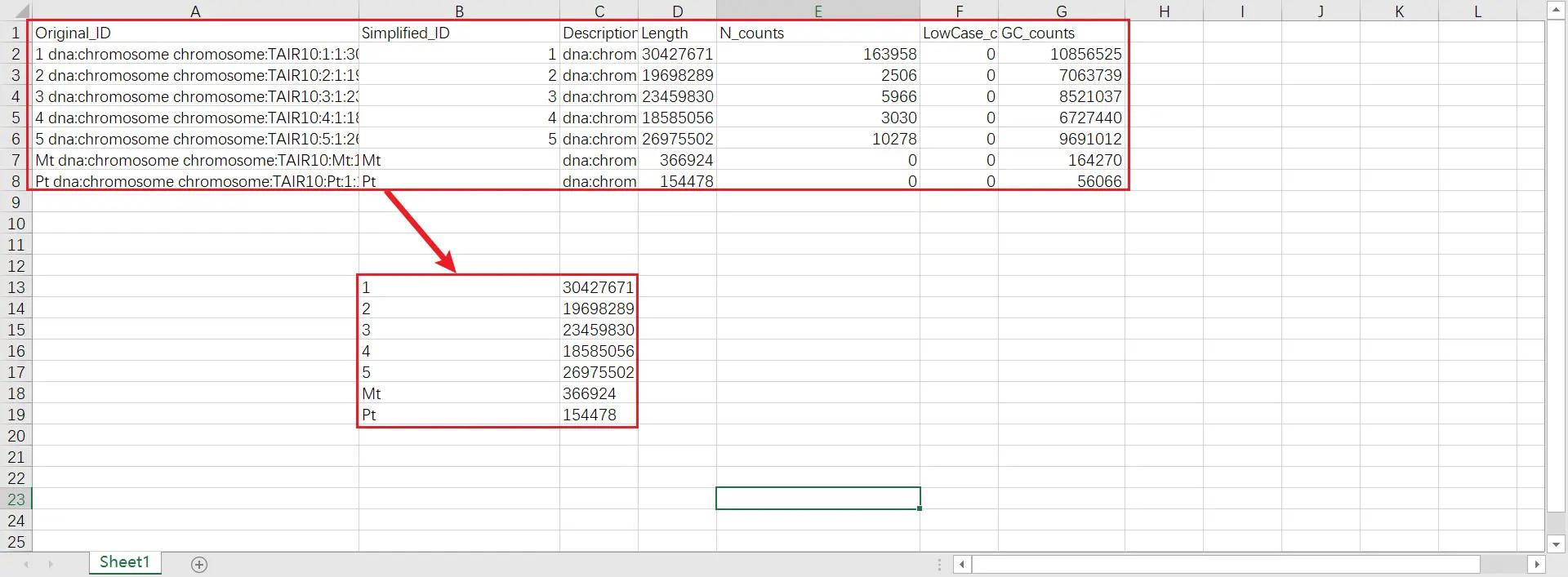
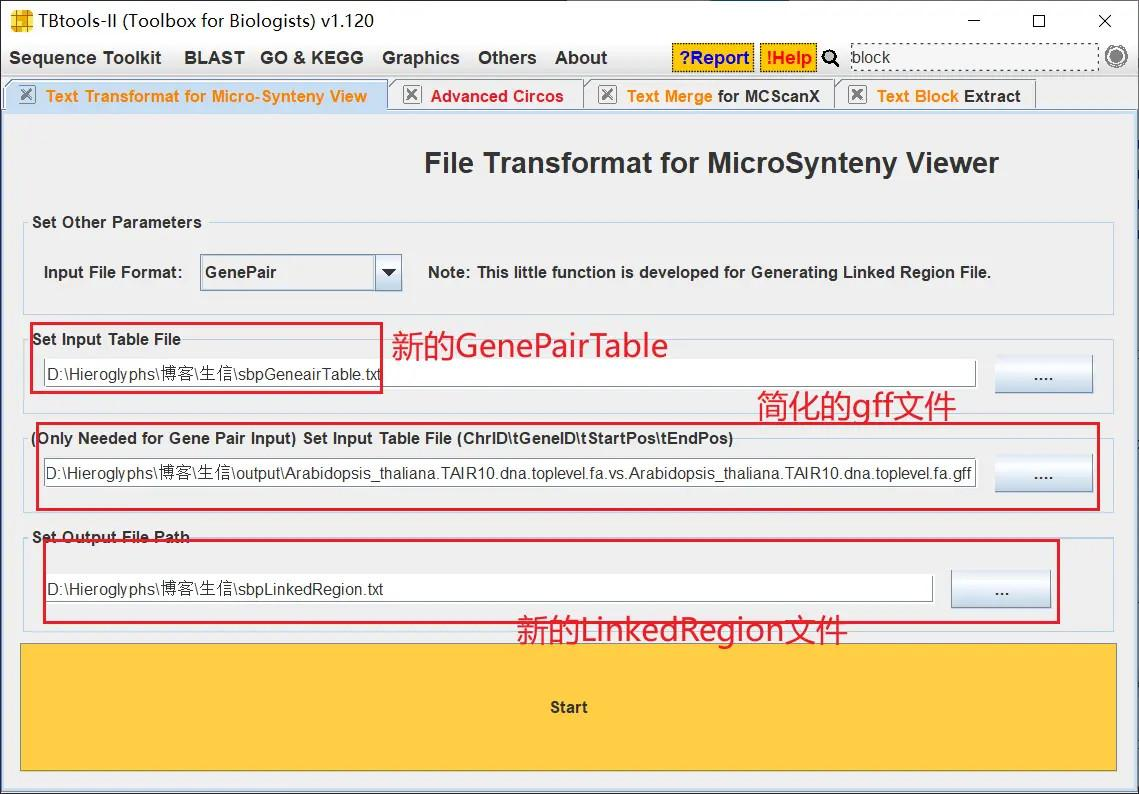
打开 TBtools 的 Text Merge for MCScanX 功能,导入上一步分析结果(在输出目录中)中的 Collinearity 文件,将分析结果转换为 GenePairTable
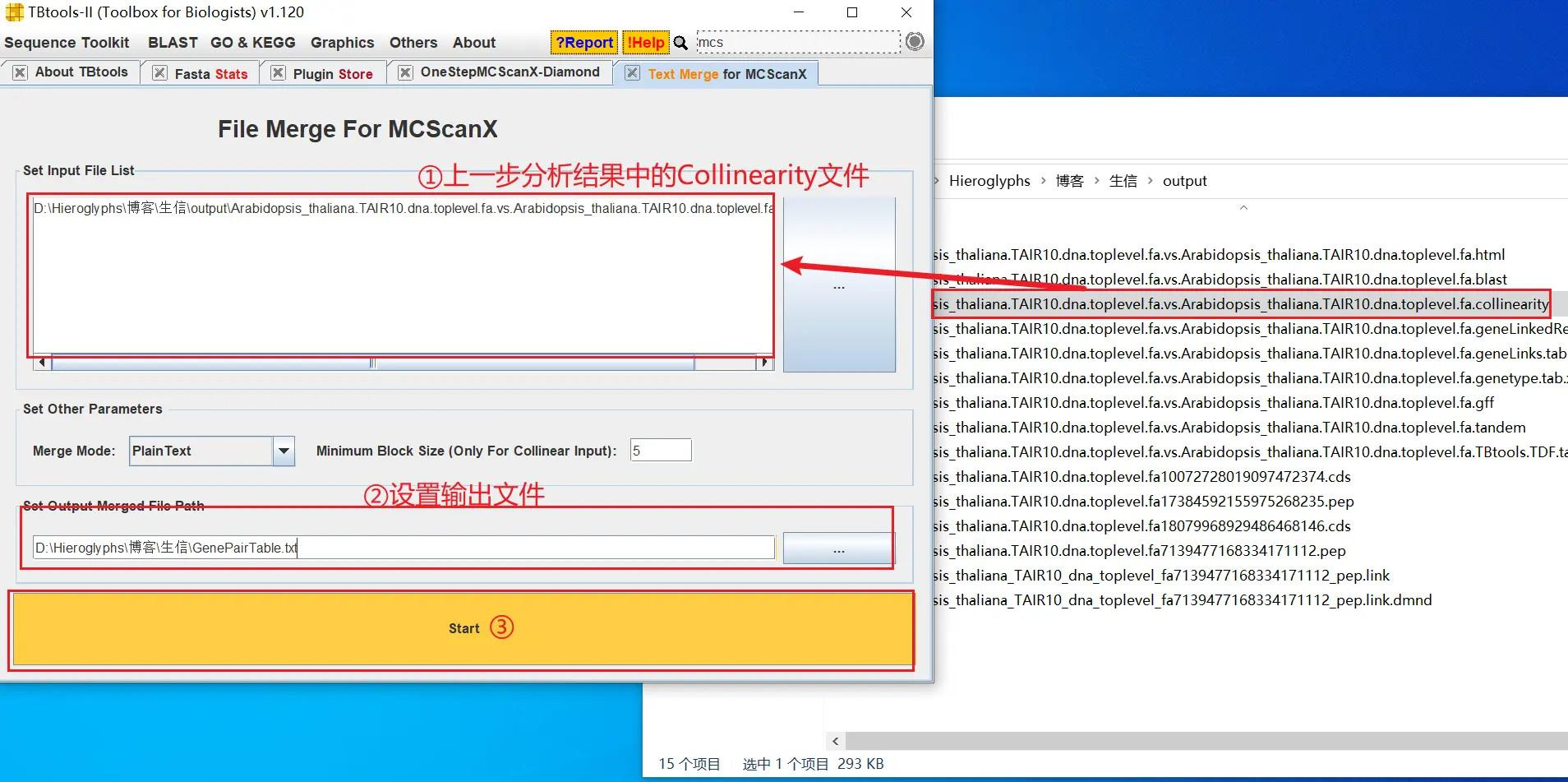
打开 TBtools 的 Text Transformat for Micro-Synteny View 功能,导入上一步获得的 GenePairTable 文件和分析结果中的以gff 为拓展名的文件(即简化后的注释文件),获得 LinkedRegion 文件,也就是基因间的关联文件 LinkedRegion
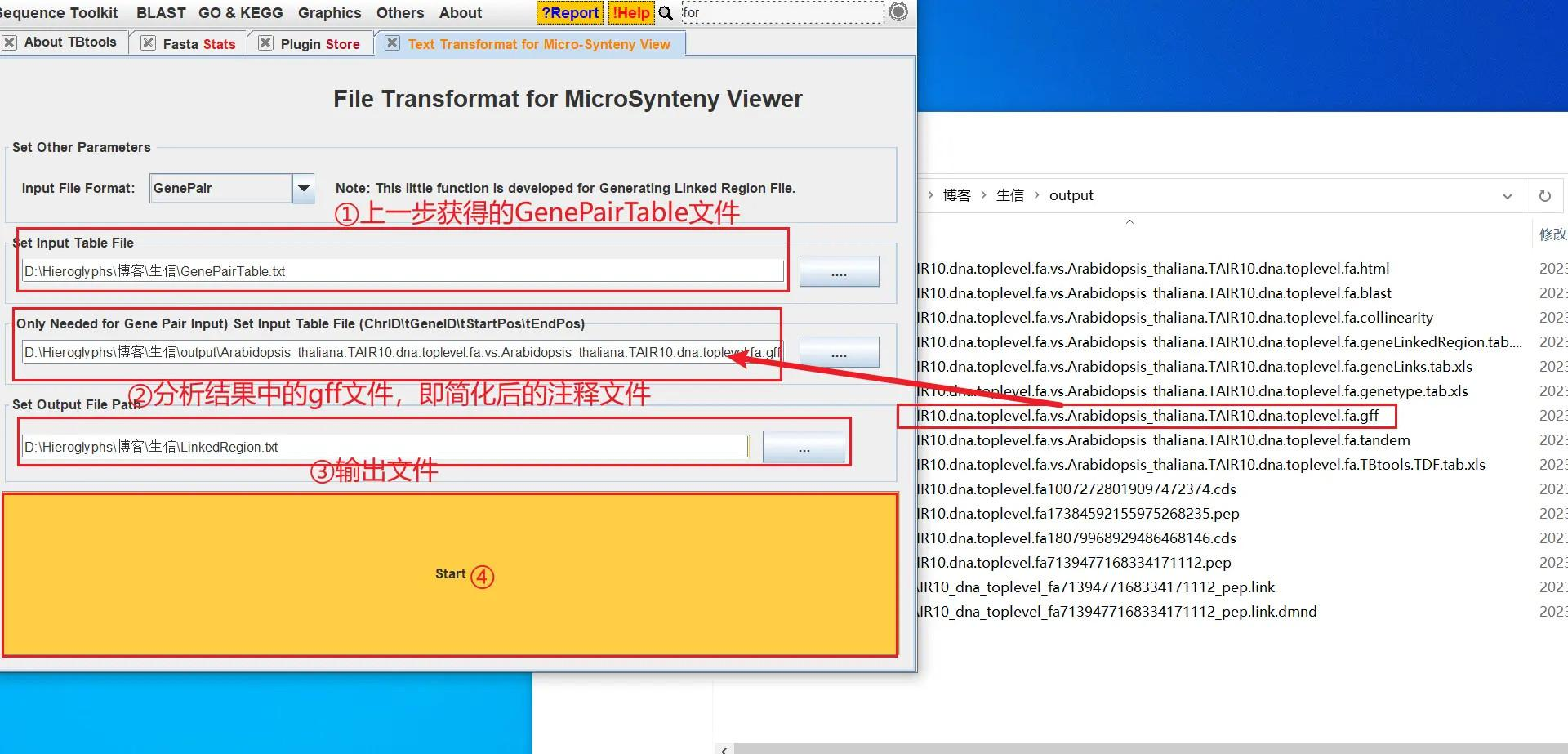
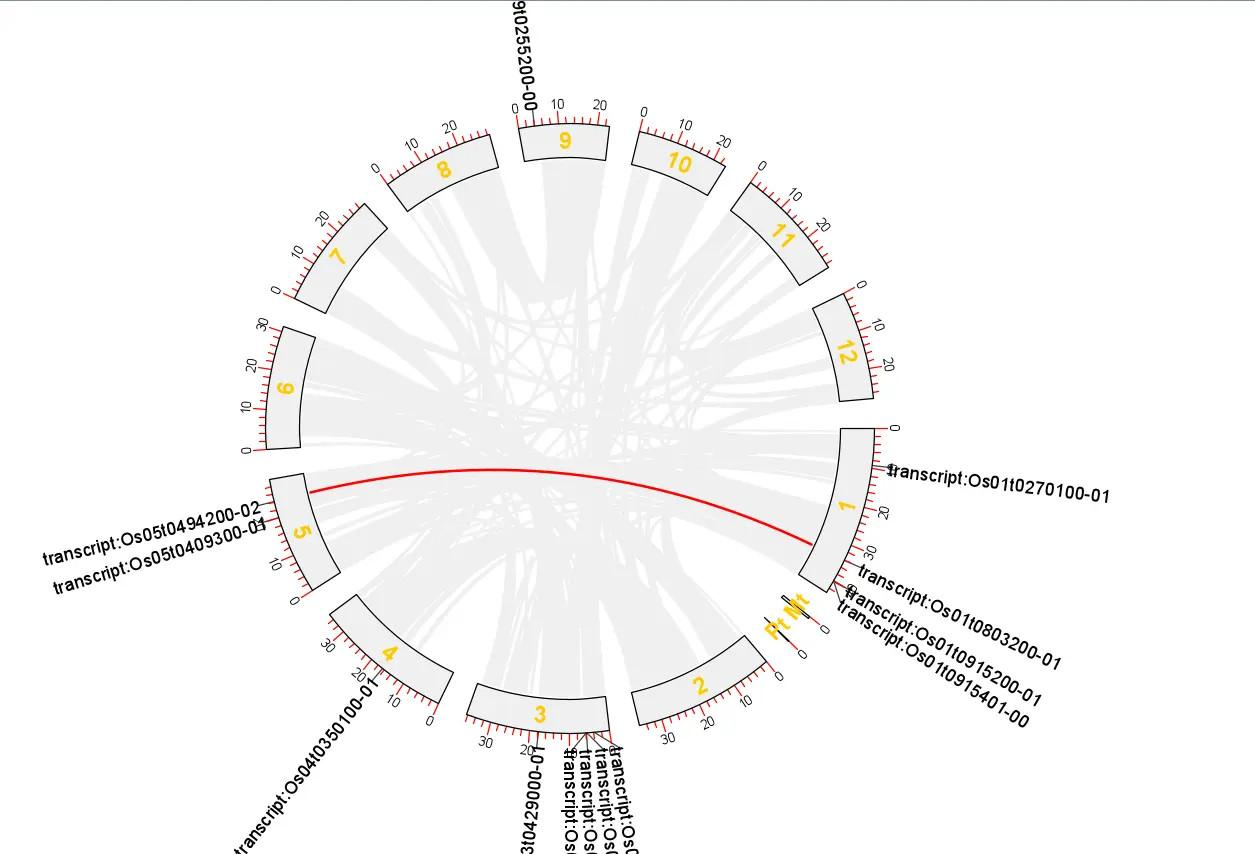
打开 TBtools 的 Advanced Circos 功能,依次导入 chr len.txt、GenePairTable.txt、LinkedRegion.txt 进行绘图
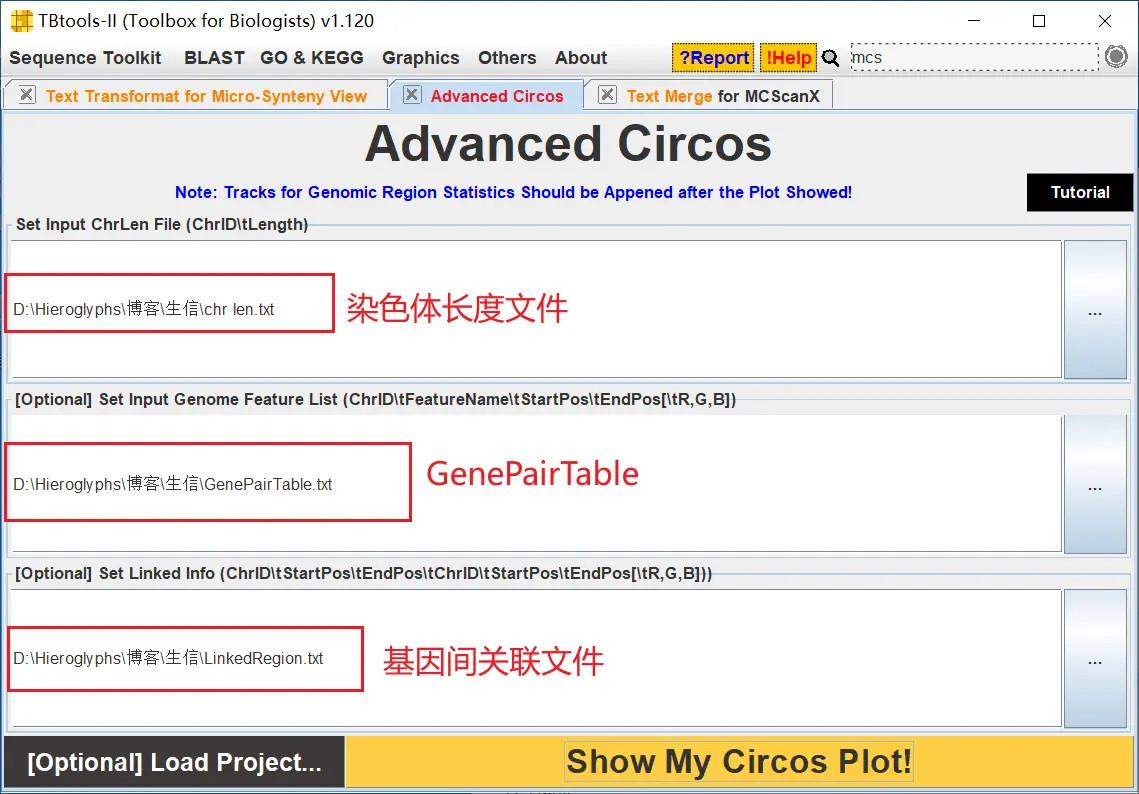
这样就得到拟南芥所有基因的共线性分析 Circos 图了
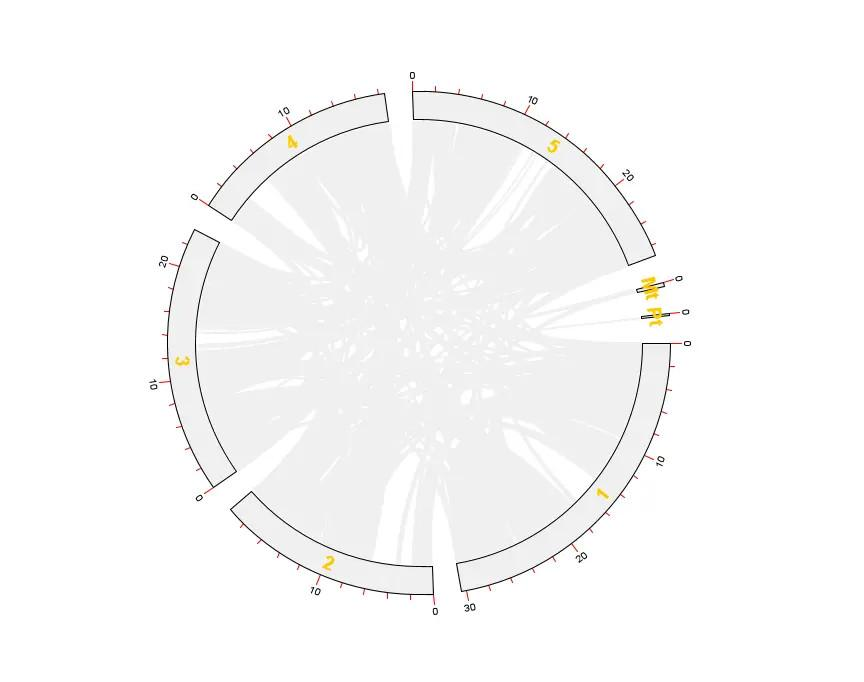
然而还没有结束,我们还需要突出显示出某些基因
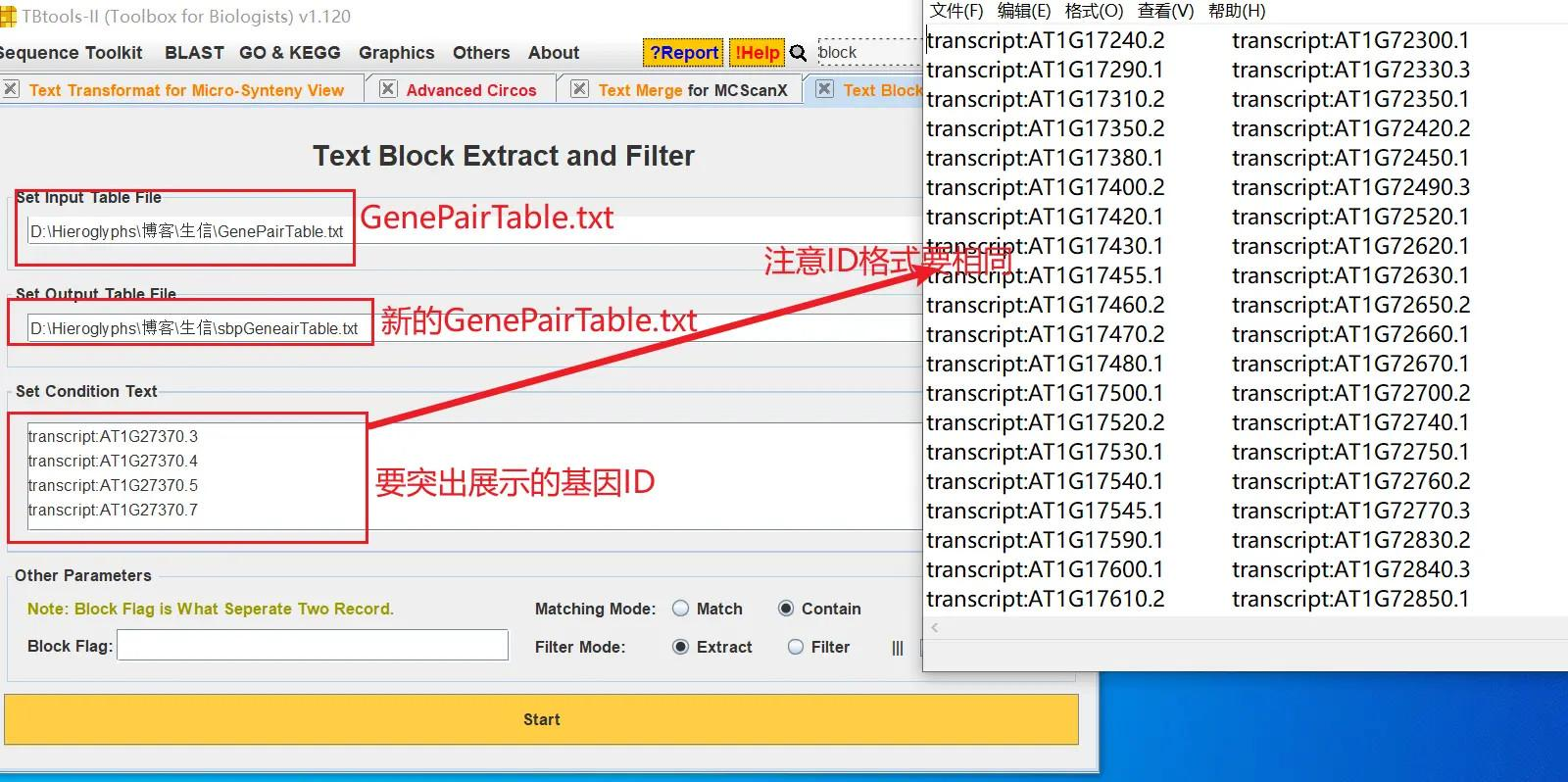
我随便抽取一些基因,打开 TBtools 的 Text Block Extract and Filter 功能,从 GenePairTable 文件中获得这些基因的对应关系,生成新的 GenePairTable 文件

然后生成新的关联文件 LinkedRegion
然后将新的 LinkedRegion.txt 文件的内容复制到 Excel 中,最后一列加上 RGB 色值

随后将 Excel 中的内容合并到之前的 LinkedRegion.txt 文件中或者新建一个文本文件保存,如果不想显示全部基因的共线性,仅仅显示选中的这些基因的共线性的话,可以直接使用刚刚保存的带有 RGB 色值的 LinkedRegion.txt 文件

我们似乎还需要显示出一些基因的名称,那就补充吧
打开 TBtools 的 Table Row Extract or Filter 功能,导入分析结果中简化的注释文件,在弹出的窗口中选择 ID 这一列,在输入框中再输入要展示的基因的 ID(注意名称要对应),这里是需要生成一个用于展示 ID 的新的 GenePairTable 文件

最后依次输入染色体长度文件、展示 ID 的 GenePairTable 文件、带有 RGB 色值的 LinkedRegion.txt 文件 绘图即可

最终结果

这里需要注意的是,TBtools 自带的 One Step MCScanX 功能分析很慢,我第一次分析水稻的共线性大概跑了四十分钟左右,这里我们可以使用 One Step MCScanX 的增强版插件,按照如下步骤进行安装
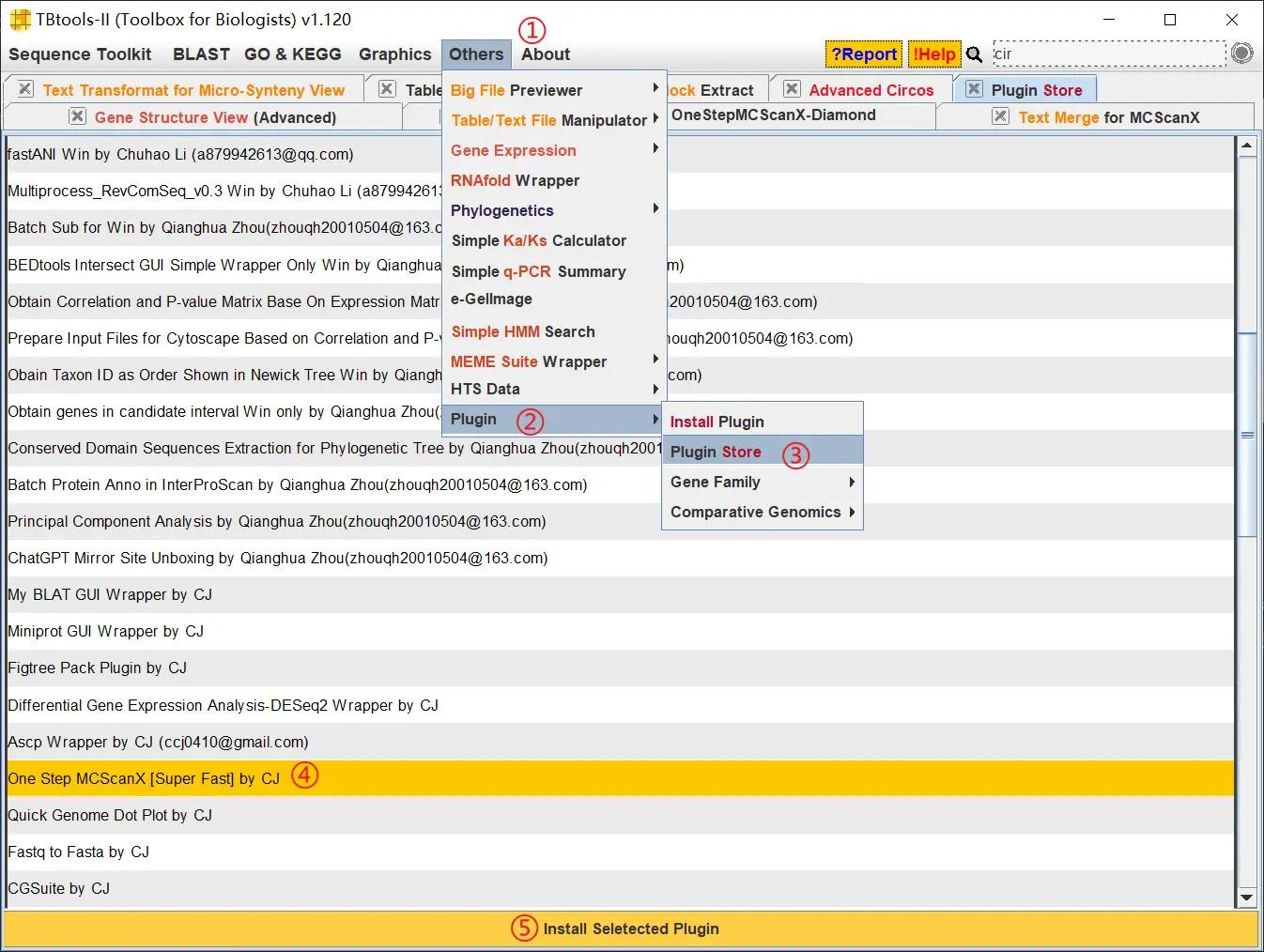
使用方法和之前的插件一样,这个插件跑一次整个水稻的共线性分析只需要几分钟
最终我绘制的图像是这样的
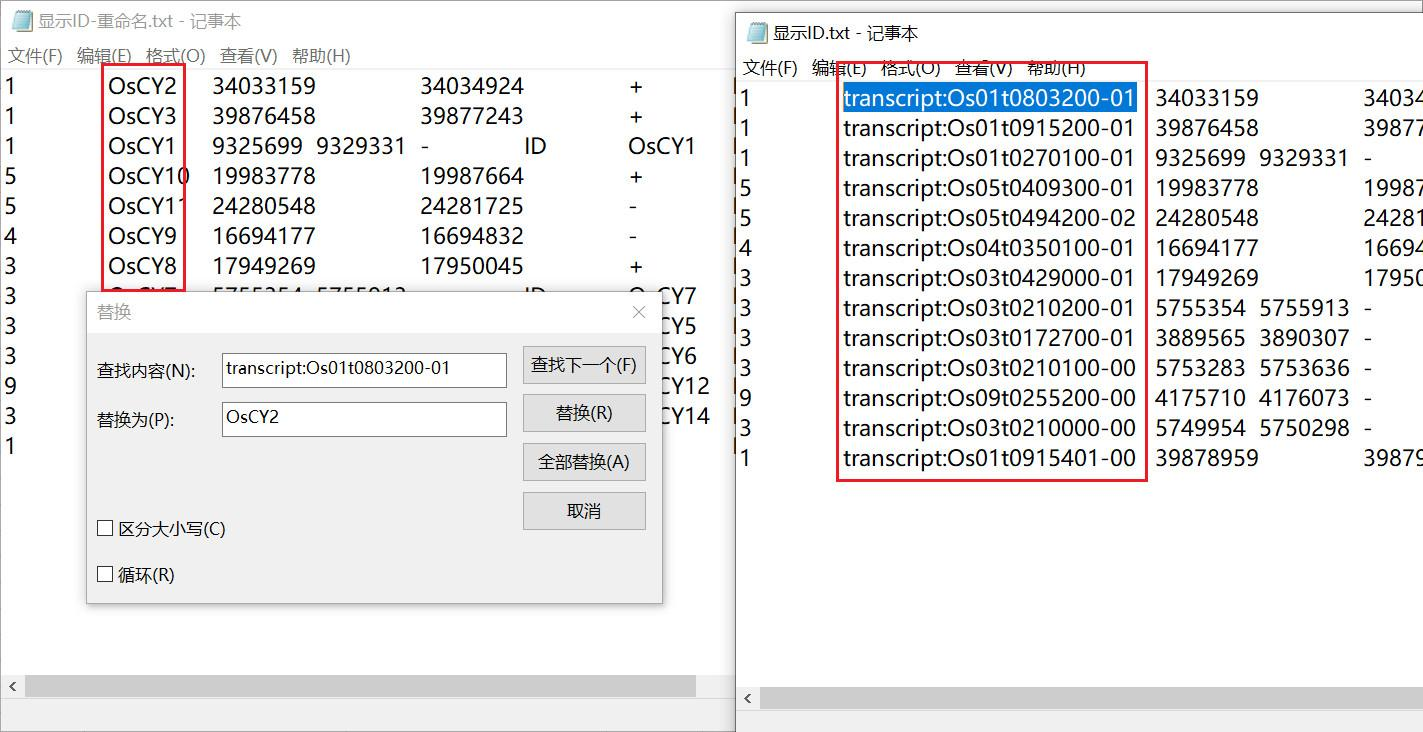
我们还是希望使用自己命名的基因名称,对显示 ID 文件中的信息进行修改,使用记事本的查找替换功能将文件中转录本的 ID 全部替换为重命名的名称即可
我们再对其进行一些美化,具体操作请参照 共线性分析美化
下图是我最终美化好的

可以看出 Cystatin 基因家族的共线性一般
多图结合
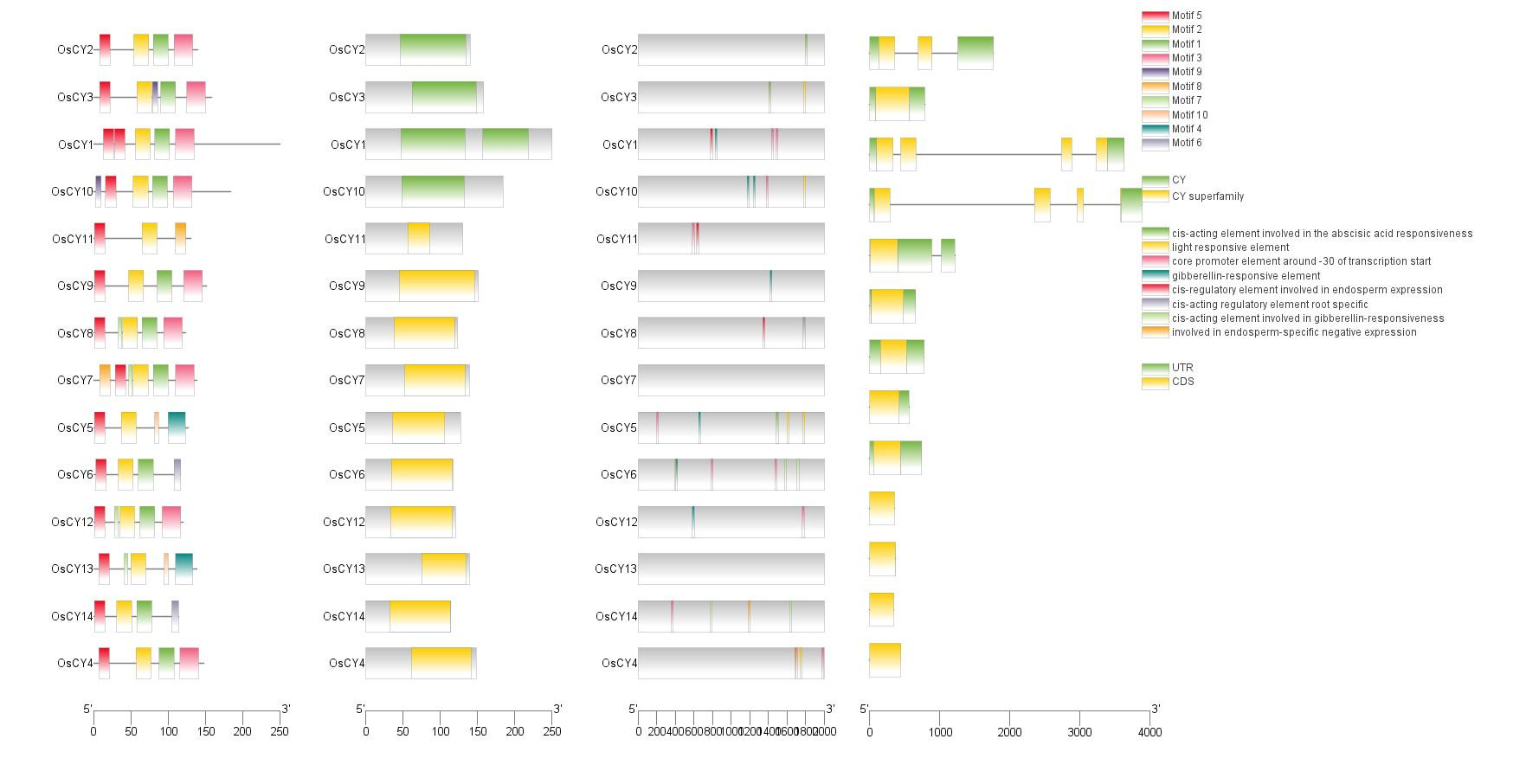
有的时候我们需要将顺式作用元件、进化树、Motif、Domain 做在同一张图上以便更直观的查看基因家族的差异,使用之前做过的这些分析的源文件将其全部整合在一张图中
打开 TBtools 的 Gene Structure View 功能,依次导入这些分析的文件进行绘图
需要注意的是,我们并不需要把这些部分全部做出来,可以根据自己的需要进行删减,想要哪几部分就对应导入那几部分的文件即可
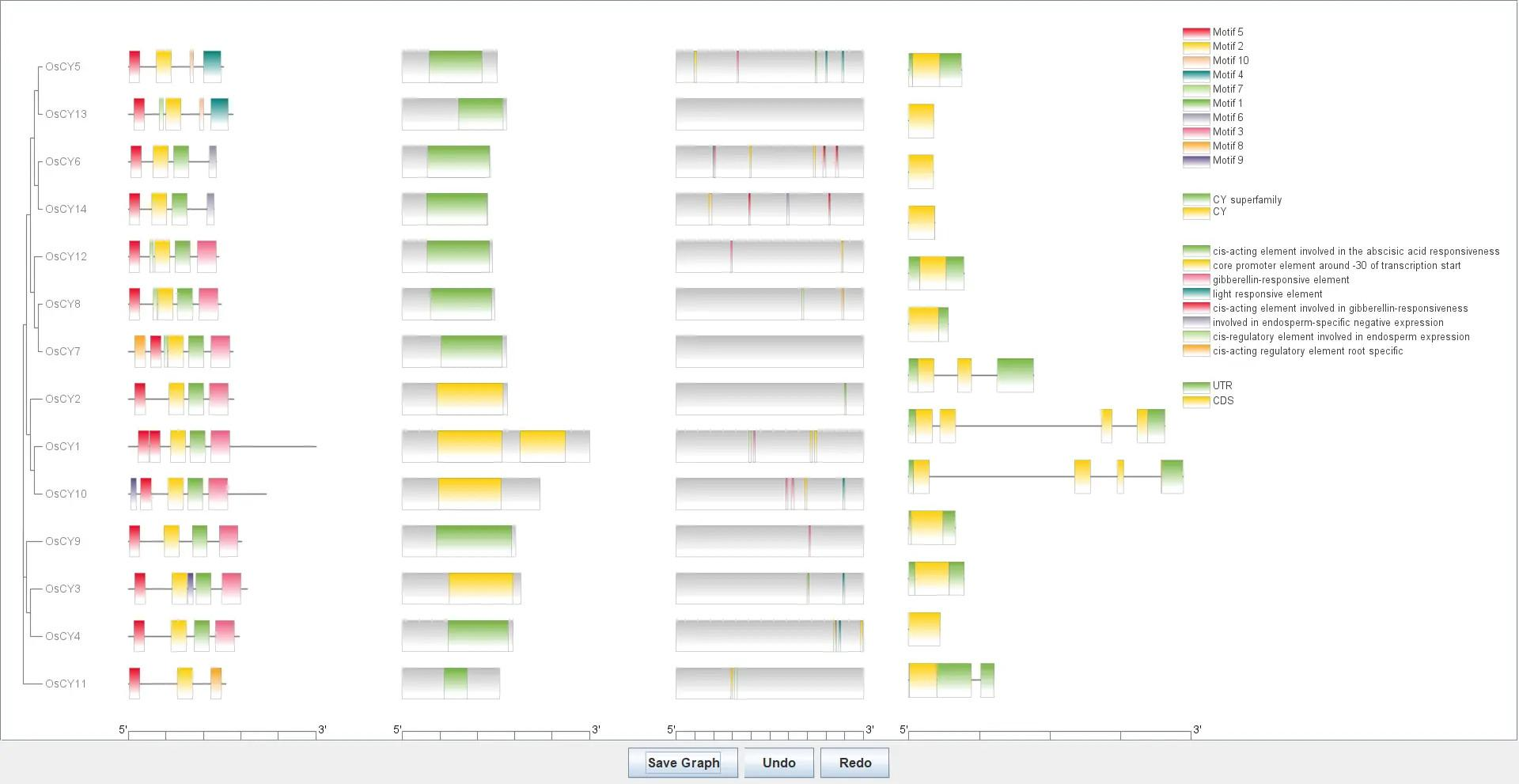
进化树+Motif

进化树+Motif+Domain

进化树+Motif+Domain+显示外显子、内含子

进化树+Motif+Domain+显示外显子、内含子+顺式作用元件
最终得到一张信息整合的图
这里必须要注意的问题就是关于每个文件中的基因名称格式,必须统一使用一样的格式才可以,任何一个文件的名称格式对不上 TBtools 都会报错,比如我们这里导入了水稻的注释文件,而水稻注释文件中的名称是 transcript:Os01t0803200-01 这样的,那么其它文件中的基因名称需要全部改为这一名称

如果想对基因进行重命名,准备好重命名文件,文件格式与染色体定位分析时的一样
在下列位置导入重命名文件
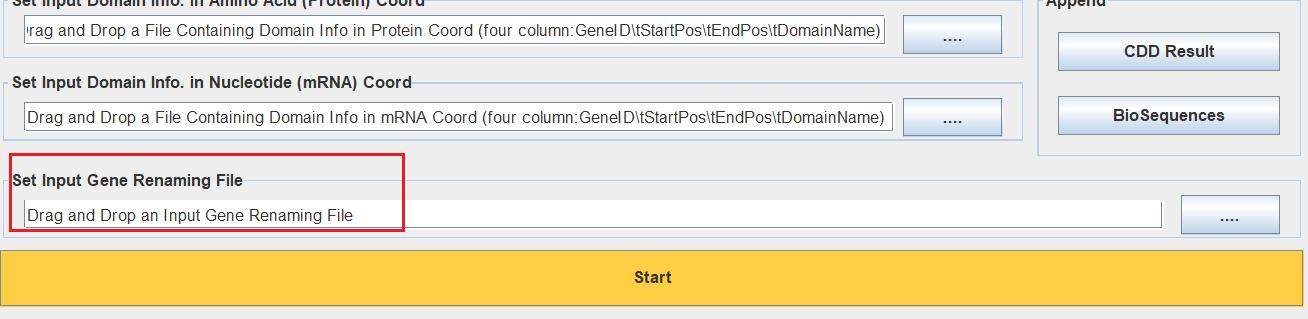
另外,如果我们不要进化树,比如在基因很少或只有单条基因的时候,可能无法做出进化树或者进化树很稀疏,那么我们可以不用导入进化树,在 Gene Structure View 功能填写进化树信息的地方我们填入基因列表即可

得出图像
跟前文染色体定位分析时一样,我们可以对图像进行一些更改,这个比较复杂,具体操作请参照 TBtools 美化 进行操作
基因家族的功能研究
功能富集分析是将基因或者蛋白列表分成多个部分,即将一堆基因进行分类,而这里的分类标准往往是按照基因的功能来限定的。换句话说,就是把一个基因列表中,具有相似功能的基因放到一起,并和生物学表型关联起来。
为了解决将基因按照功能进行分类的问题,科学家们开发了很多基因功能注释数据库。这其中比较有名的一个就是 Gene Ontology(基因本体论,GO)和 Kyoto Encyclopedia of Genes and Genomes(京都基因与基因组百科全书,KEGG)。
其中,GO 是基因本体论联合会建立的一个数据库,旨在建立一个适用于各种物种的、对基因和蛋白功能进行限定和描述的,并能随着研究不断深入而更新的语义词汇标准。GO 注释分为三大类,分别是:分子生物学功能(Molecular Function,MF)、生物学过程(Biological Process,BP)和细胞学组分(Cellular Components,CC),通过这三个功能大类,对一个基因的功能进行多方面的限定和描述。
而 KEGG,大多数听说过 KEGG 的人都会把它当做一个基因通路(Pathway)的数据库,其实人家的功能远不止于此。KEGG 是一个整合了基因组、化学和系统功能信息的综合数据库。KEGG 下属 4 个大类和 17 和子数据库,而其中有一个数据库叫做 KEGG Pathway,专门存储不同物种中基因通路的信息,也是用的最多的一个,所以,久而久之,KEGG 就被大家当做是一个通路数据库了
功能富集分析的算法有很多种,能够做功能富集分析的工具也非常多,如果大家想深入了解的话,下面是一个工具列表可供大家学习。
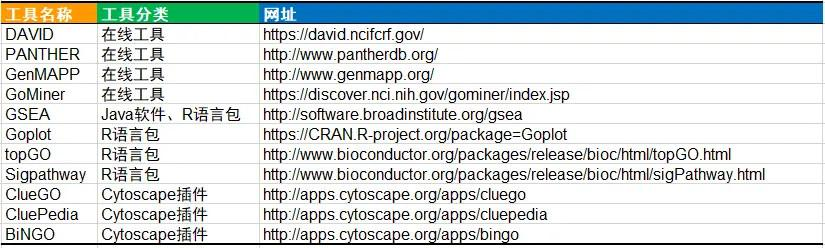
在以上所有的工具中,有一个工具是最为常用,也最为权威,那就是 DAVID,DAVID 是由美国 Leidos 生物医学研究公司的 LHRI 团队开发的一个在线基因注释及功能富集网站,其网址为 https://david.ncifcrf.gov 为什么说 DAVID 它是最权威的呢?看下图就知道了:仅 DAVID 这一个软件就发表了 10 篇 sci 文章,其中 5 分以上 7 篇,累计影响因子将近 85 分。其他用 DAVID 进行分析并发表的文章就更不计其数了
GO 富集分析

打开 DAVID 官网:https://david.ncifcrf.gov/
点击上方功能菜单 Functional Annotation
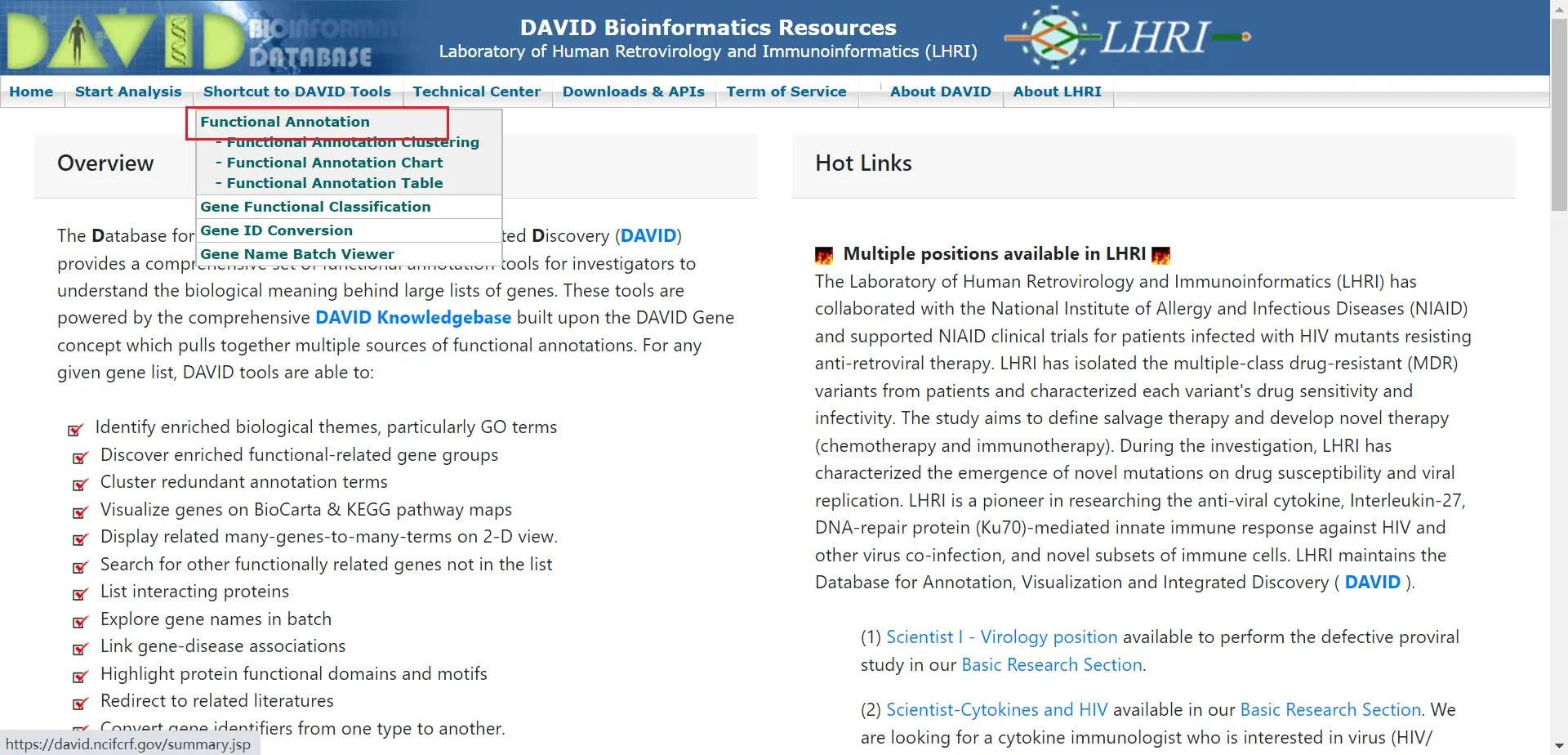
选择上方的 upload 选项卡,在编辑框内粘贴基因 ID 列表,选 ID 类型为 ENSEMBL_GENE_ID ,选择列表类型为基因列表,最后上传列表

现在展示出的即富集分析结果,取消勾选上方 Check Defaults 取消全选,展开 Gene_Ontology 项,勾选 BP、CC、MF 项,点击最下方 Functional Annotation Chart 将勾选的项目以列表形式给出

随后就会跳出一个浏览器页面,以列表的形式将上述结果展示给我们了,点击右上方 Doanload File 将新弹出窗口中的数据复制保存在 Excel 表格中,按顺序保留下列数据
此处我解释一下 BP、CC、MF 项分别为什么意思
GO 是 Gene Ontology 的缩写,是一种用于生物信息学和计算生物学中的术语表达的标准格式。GO 主要由三个部分组成:分子功能(Molecular Function,MF)、生物过程(Biological Process,BP)和细胞组分(Cellular Component,CC)这些术语描述了基因和蛋白质的不同功能和所处的环境
BP(Biological Process)是指分析对象(如基因)所参与的生物学过程,通常是一组相互作用的生物学事件,这些生物学事件在时间和空间上有一定的结构和组织,比如细胞代谢、信号转导、生物合成等等
CC(Cellular Component)是指分析对象(如蛋白质)所在的细胞组成部分,即它所在的细胞器、细胞构造和細胞組織中对应的位置。比如细胞膜、线粒体、细胞核等等
MF(Molecular Function)是指分析对象(如基因或蛋白质)具有的分子功能,即它所扮演的特定化学活性,比如催化反应、配体结合、蛋白质结合、转录因子结合等
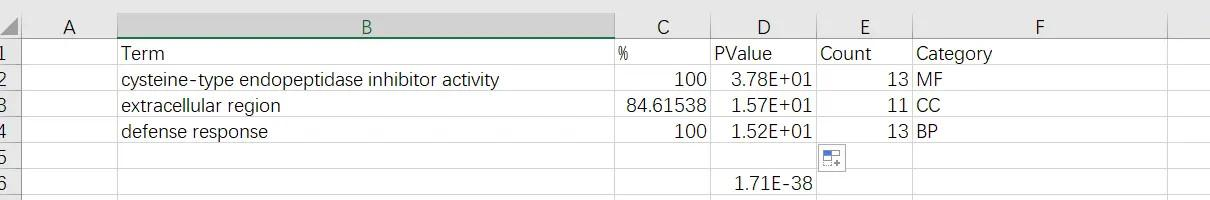
第一列:富集的名字(Term)
第二列:是 X 轴显示内容,例如 gene ratio 等(%列)
第三列:p 值或者 fdr,即图中的颜色,根据 p 值变化(p-value 列)
第四列:基因数,控制气泡大小(Count)
第五列:为可选列,为分类信息,例如 BP,CC,MF 等(Category)
在制作气泡图时,通常会对富集分析结果中的 P 值进行转换,以避免图像上 P 值的差异过分压缩的情况。常用的转换方式是将 P 值进行-log10(以对数值)的处理

对数据进行整理,简化一下名称,最终得到如下数据
打开微生信平台(http://www.bioinformatics.com.cn/)的富集气泡图功能进行绘图,也可以使用 R 和 Excel,R 的环境比较复杂,这里我就不放代码了。

在这个 GO 富集分析结果中,我们可以从不同的角度来分析:
在功能方面(GOTERM_MF_DIRECT),我们可以看到,13 个基因处于“半胱氨酸型内切酶抑制剂活性”的功能位置,这一结果的贡献值(P 值,Bonferroni、Benjamin 人为误判率)非常小,表明这 13 个基因的相似分布可能不是随机事件造成的,进而推断这些基因在相关的生物过程、疾病或免疫应答方面扮演着重要的作用
在细胞组分方面(GOTERM_CC_DIRECT),这 13 个基因中有 11 个基因分布于“细胞外区域”,并具有非常高的显著水平和 FDR 调整系数值,表明这些基因在“细胞外区域”方面的富集并不是随机发生的,这表明这些基因可能参与了水稻的细胞外相关生理作用和功能
在生物过程方面(GOTERM_BP_DIRECT),这 13 个基因中的所有基因均涉及到“防御反应”,且 P 值和调整后的 P 值均非常小,这表明这些基因在水稻防御反应中可能扮演一个重要的角色
因此,结合这些结果,这些基因群应该与防御反应有关,它们在水稻中可能扮演着一种重要的保护性功能
KEGG 富集分析
还未完成
总结
本文原本是参考文献《拟南芥和水稻 Cystatin 基因家族的生物信息学分析_杨泽峰》知网链接 中的 Cystatin 基因家族来做的,但是最后又发现了 2 个 Cystatin 基因家族的基因。以上便是基因家族鉴定的总体流程。