文献阅读与复现|磷酸盐饥饿反应调节受体样激酶 OsADK1 是菌根共生和磷酸盐饥饿反应所必需的
本文总阅读量 次
文章题为"A phosphate starvation response-regulated receptor-like kinase, OsADK1, is required for mycorrhizal symbiosis and phosphate starvation responses"。该文章发表于 New Phytologist(中科院一区,IF=9.3,第一作者:Jincai Shi,通讯作者:Ertao Wang)。
文献内容简介
在植物与丛枝菌根真菌(Arbuscular mycorrhizae,AM)之间建立的共生关系中,丛枝菌根真菌通过形成树突状结构的树突状体在植物根内皮质细胞内促进与宿主植物的营养物质交换。在这种共生过程中,植物根释放出异戊烯内酯刺激丛枝菌根真菌的代谢和分枝。同时,丛枝菌根真菌也释放出一些真菌信号,包括壳寡糖和脂质壳寡糖,这些信号被水稻中的 LysM 受体样激酶复合物识别并引发核内的钙振荡反应。通过这一信号传导通路,一系列 AM 共生中的特异转录因子的调节被调控,如 RAM1、RAD1、DIP1 等,这些因子影响树突状体的形态和发育以及根皮质细胞的营养物质交换。此外,植物磷缺乏响应相关基因 OsPHR1/2/3 在 AM 共生中也发挥关键作用,可以直接激活异戊烯内酯生物合成相关基因和 PAM 特异转运蛋白基因等相关基因,以调节植物对磷的吸收和利用。本文作者通过转录组深度测序和 DNA 亲和纯化测序分析,发现了 OsPHR1/2/3 的靶基因 Arbuscular Development Kinase 1(OsADK1),并证实了在 AM 共生中 OsPHR2 可以直接调控 OsADK1 的表达,从而影响树突状体的发育过程。
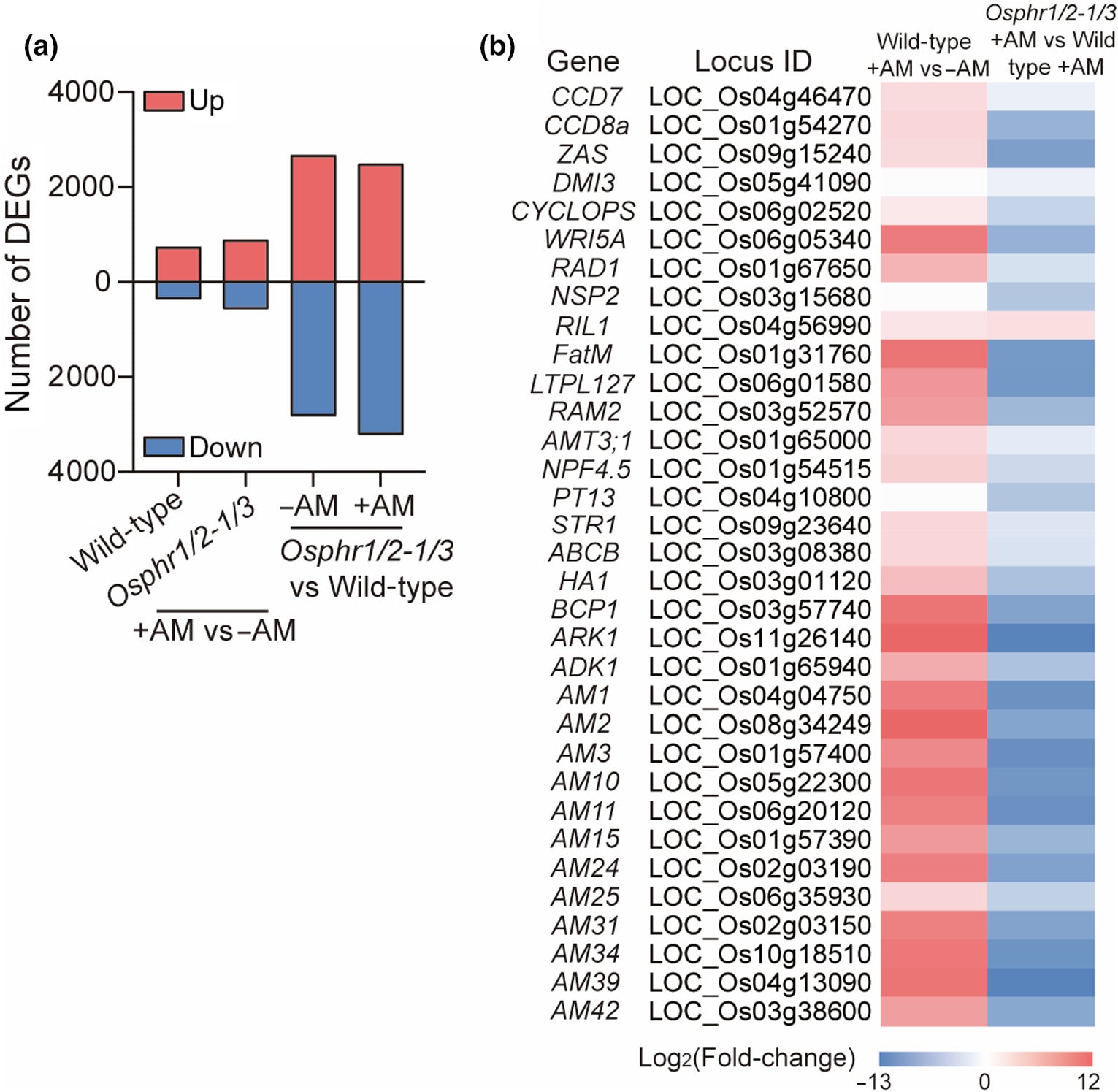
为了研究 OsPHR1/2/3 在 AM 共生中的作用,作者对定植和非定植的野生型水稻植株以及 OsPHR1/2/3 三重突变体水稻植株进行了转录组测序。通过对测序数据进行分析,在 AM 定植的野生型植株中发现了 1106 个差异表达基因;在突变体植株中发现了 1464 个差异表达基因。对 AM 定植和非定植的野生型植株以及突变体植株的转录组进行差异表达分析,发现有 5716 个(AM 定植)和 5499 个(非定植)差异表达基因。大部分 AM 调节基因在 Osphr1 中相对于野生型的突变体植株表达差异较大。
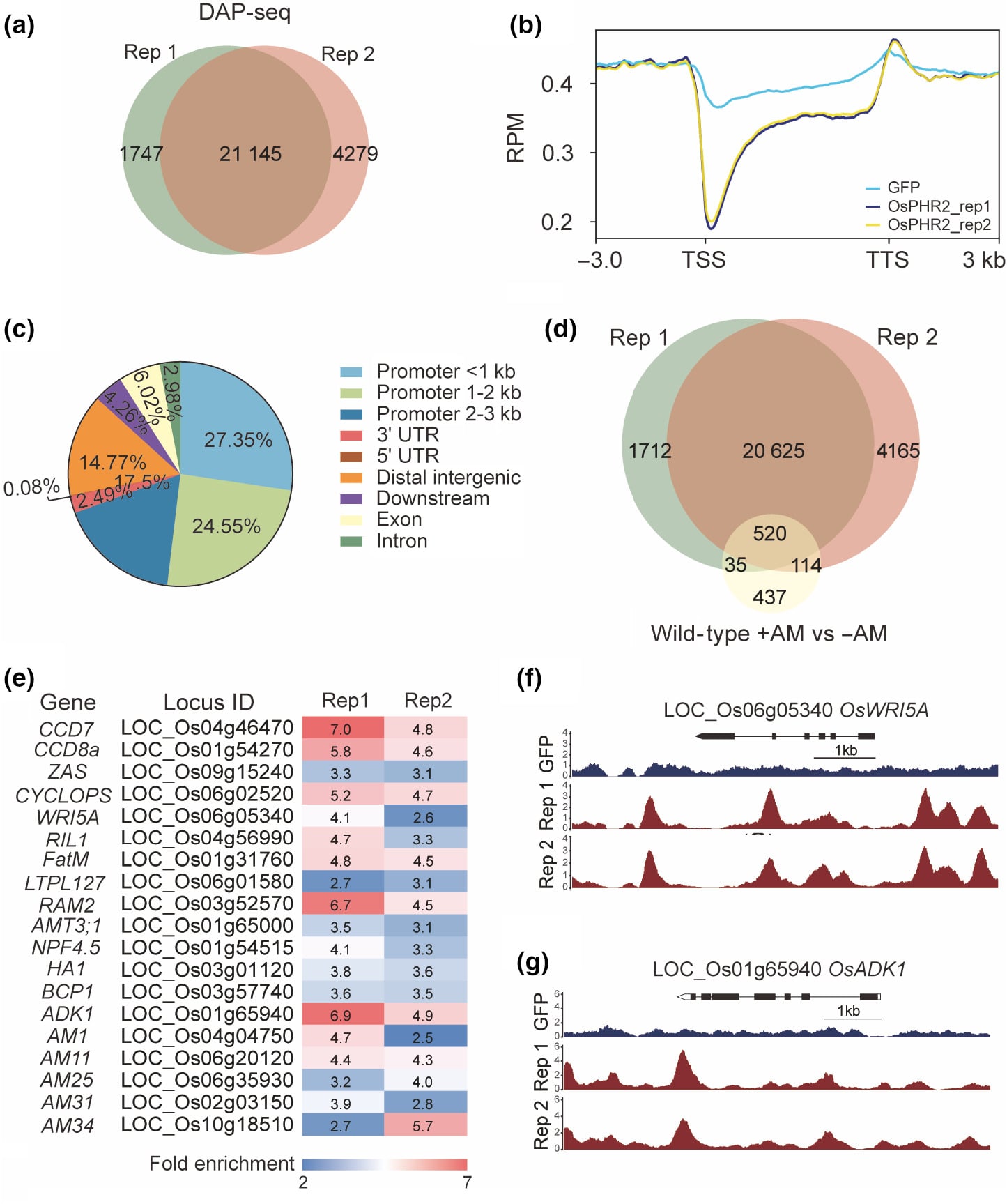
DAP-seq 分析结果表明,OsPHR2 结合位点富集于靶基因的转录起始位点附近(图 2a),主要分布在远端基因间和外显子区域(图 2c),这表明 OsPHR2 结合位点在启动子区域有显著富集特征。同时,在 OsWRI5A 的启动子区域检测到强的 OsPHR2 结合峰(图 2d)。这些结果表明 OsPHR2 可能在靶基因的转录调控中起着重要的作用,其可能通过结合到启动子区域来影响靶基因的转录起始。
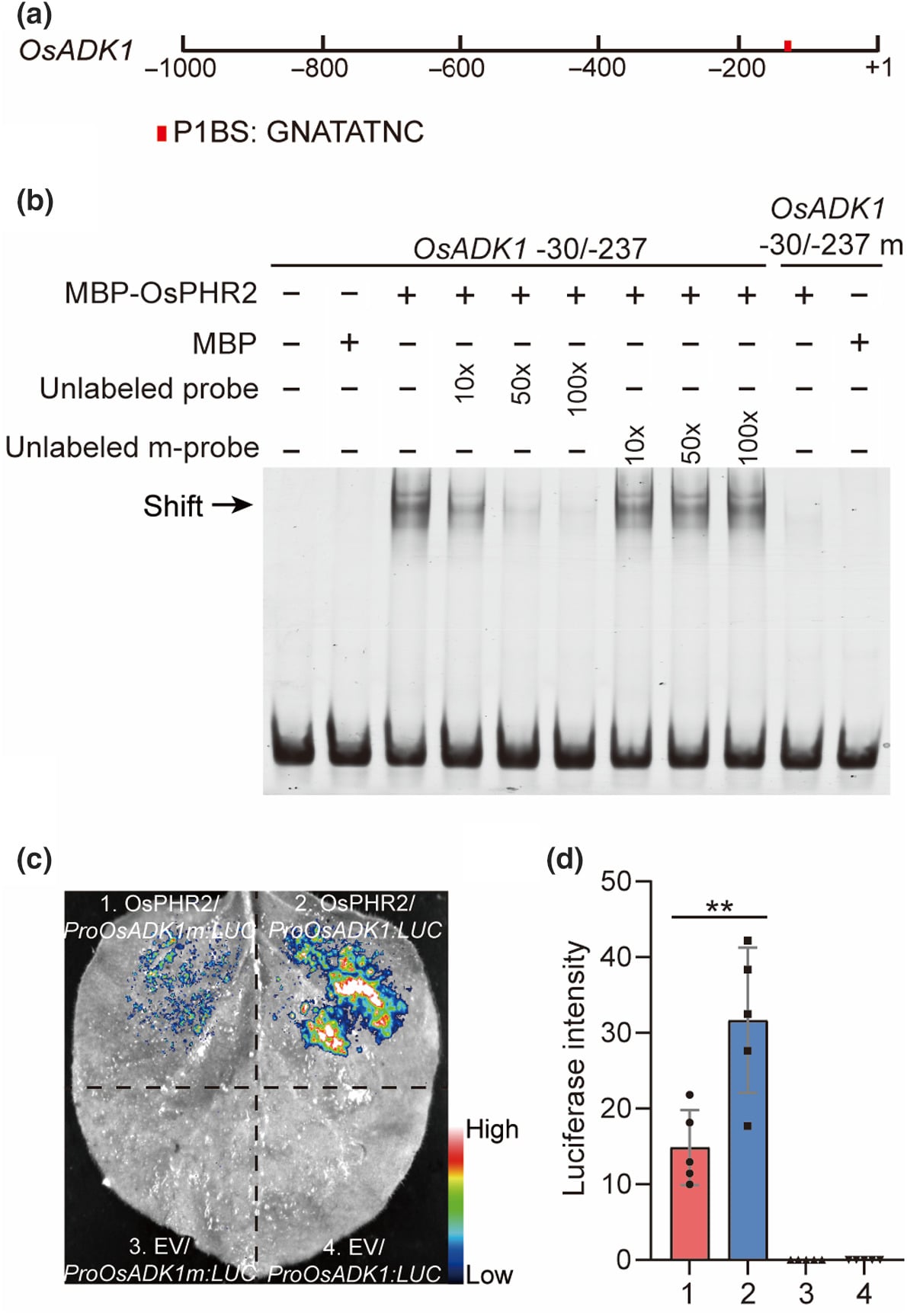
作者在水稻 OsADK1 基因的启动子区域中识别出了与 OsPHR2 蛋白结合的 P1BS 元件(图 3a)。为验证 OsPHR2 是否能直接与 OsADK1 启动子结合,作者通过凝胶迁移率变动分析(EMSA)测试了一个包含 P1BS 基序及 MBP 融合蛋白的 OsADK1 启动子片段。实验结果表明,OsPHR2 能够结合含有 P1BS 的启动子片段,却不能与突变的 P1BS 序列结合(图 3b),说明 OsPHR2 具有特异性结合 P1BS 元件的能力。为进一步确认 OsPHR2 是否能促进植物中 OsADK1 的表达,作者还在烟草叶片中进行了双荧光素酶报告实验。实验中将 OsADK1 启动子控制的荧光素酶报告基因与 35S 启动子驱动的 OsPHR2 表达载体进行共渗透,并以空载体作为阴性对照。实验结果显示,35S 启动子驱动的 OsPHR2 表达载体显著提高了荧光素酶活性,而空载体对照组无此现象(图 3c,d),这表明 OsPHR2 能够激活植物中 OsADK1 的转录。
OsADK1 编码的蛋白质由 637 个氨基酸组成,属于不含跨膜域的细胞质受体样激酶,具备酪氨酸激酶结构域。为验证其功能,作者在大肠杆菌中表达并纯化了带有 His 标签的 OsADK1(HIS-TF-OsADK1)。实验结果证实 HIS-TF-OsADK1 具有自磷酸化功能(图 4a),符合其作为受体样激酶的特性。系统发育分析表明,OsADK1 存在于菌根植物的基因组中,但不存在于括拟南芥在一类的非菌根植物(图 4b),这表明了 OsADK1 在 AM 共生中的作用。通过 qRT-PCR 实验,作者测定了菌根定植植株与野生型植株根部 OsADK1 的表达水平,结果显示,菌根定植的根部 OsADK1 表达量显著提高,并随接种时间延长而进一步增加(图 4c,d)。此外,研究还发现菌根共生过程中 OsADK1 的表达诱导依赖于 OsPHR1/2/3(图 4e,f)。
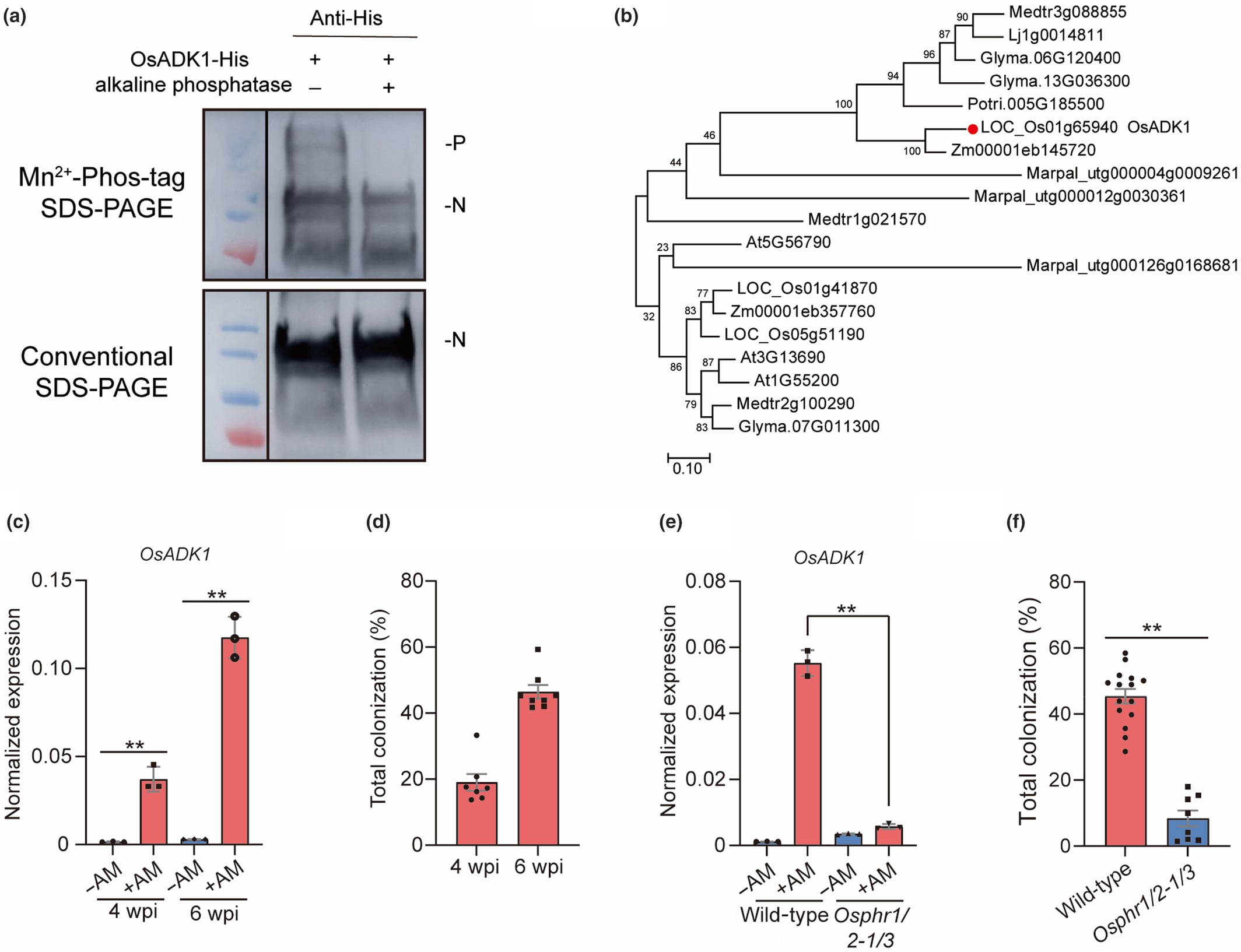
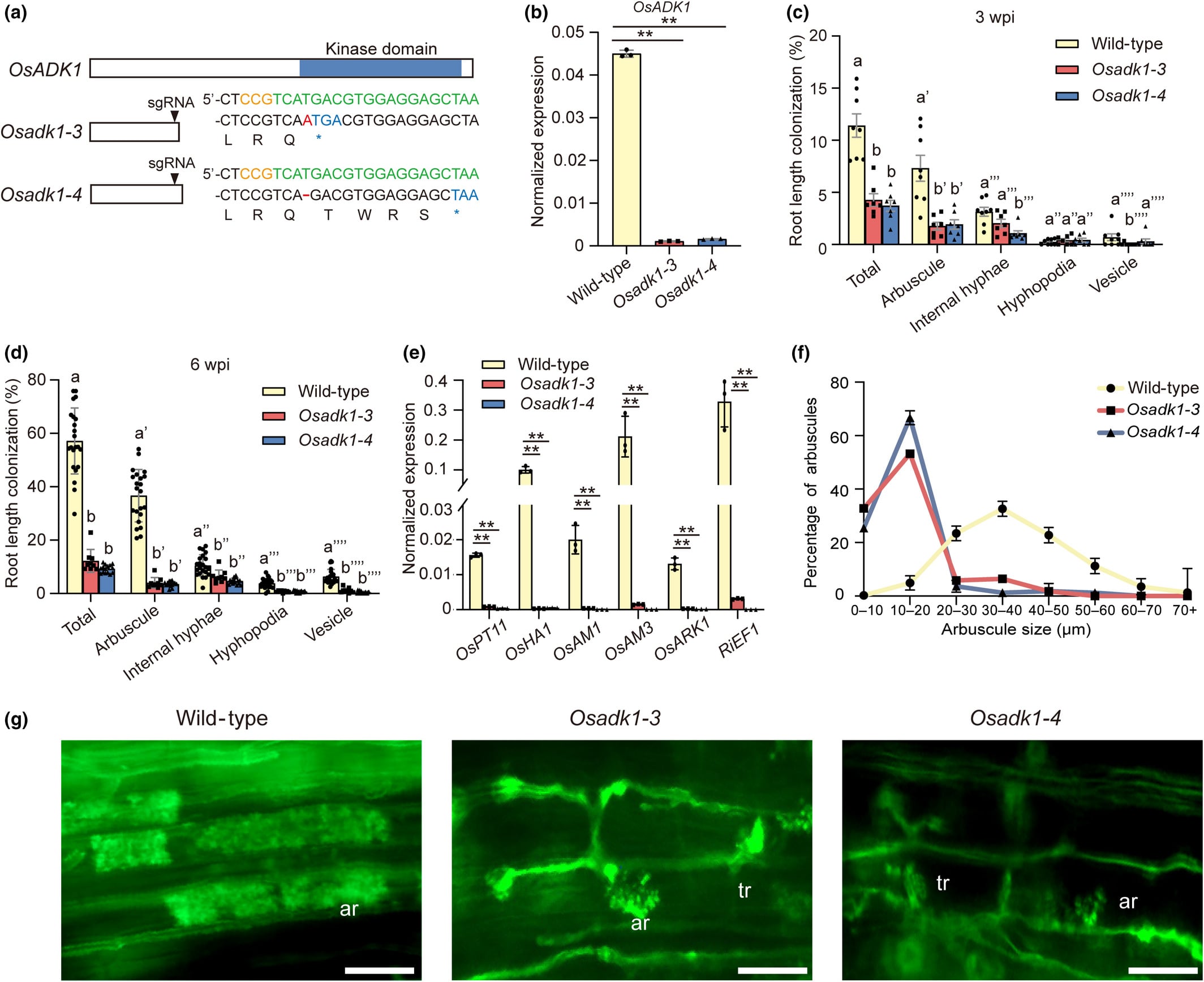
为探究 OsADK1 在菌根共生中的作用,作者通过 CRISPR 基因组编辑技术成功构建了两个 OsADK1 敲除突变体,并从中鉴定出两个新的 OsADK1 突变等位基因(Osadk1-3 和 Osadk1-4)。这两个等位基因在 OsADK1 编码区第 444 个核苷酸位置分别发生 1bp 的插入和缺失,导致移码和提前终止翻译(图 5a)。与野生型植株相比,突变体中 OsADK1 的表达显著降低(图 5b)。通过对 Osadk1-3 和 Osadk1-4 突变体植株的菌根表型及形成情况进行观察发现,与野生型相比,OsADK1 突变体在菌根定植方面展现出显著的缺陷,并且在丝足形成以及皮质内菌丝的侵染程度方面也均低于野生型(图 5c,d)。此外,在 Osadk1 突变体植株中,共生标记基因(包括 OsAM1、OsAM3、OsPT11、OsHA1 和 OsARK1)以及真菌基因 RiEF1 的转录水平显著低于野生型(图 5e)。对丛枝大小的分析发现,野生型植株的丛枝大小主要集中在 30-40 微米,而 Osadk1 突变体中 10-20 微米的小丛枝比例有所增加(图 5f,g),这表明 OsADK1 在丛枝发育过程中扮演着关键角色。
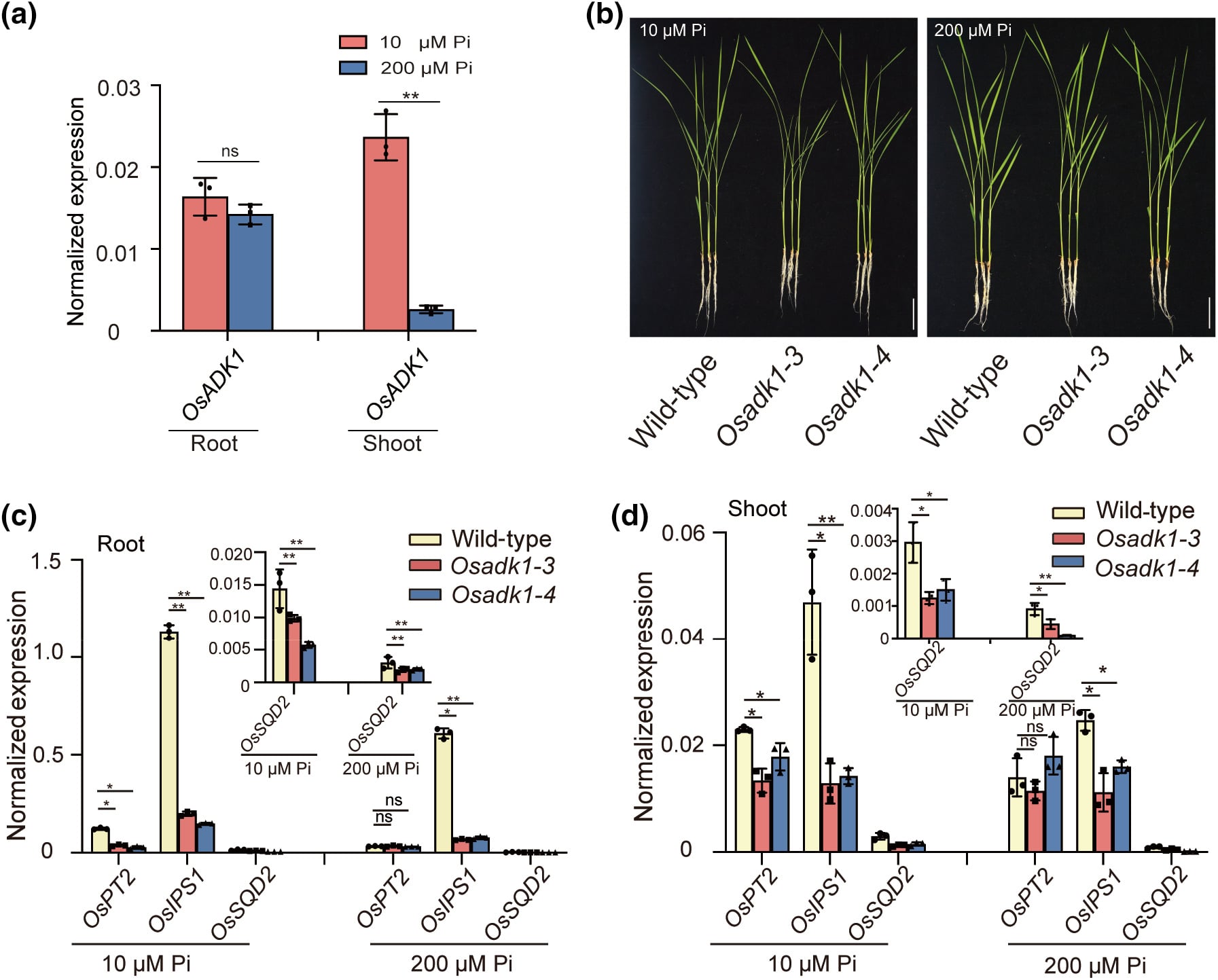
由于 OsADK1 直接受到 OsPHR2 的调控,作者进一步探讨了 OsADK1 在植物磷(Pi)饥饿反应中的作用。在磷缺乏(10μM)和磷充足(200μM)的条件下,OsADK1 在根部的表达保持较低水平,但在磷缺乏条件下,其在芽部的表达显著上调(图 6a)。与野生型相比,在磷缺乏和磷充足条件下,Osadk1 突变体中磷饥饿响应基因 OsSQD2、OsPT2 和 OsIPS1 的表达均显著降低(图 6c,d),这说明 OsADK1 参与调节了磷饥饿反应相关基因的表达。然而,无论是磷缺乏还是磷充足条件下,Osadk1 突变体的植物生长并未受到影响(图 6b)。
文献分析与总结
本研究通过转录组深度测序和 DNA 亲和纯化测序,鉴定了水稻转录因子 OsPHR1/2/3 在 AM 共生过程中的 520 个直接靶标基因。这些基因涉及内酯合成、转录重编程和双向养分交换。另外,研究还发现了一个新的目标基因,即丛枝发育激酶 1(OsADK1),它是 OsPHR1/2/3 的目标基因之一。电泳迁移实验和转录激活实验显示,OsPHR2 能直接结合到 OsADK1 启动子中的 P1BS 元件,并启动其转录。OsADK1 对于菌根结构的建立和丛枝发育至关重要。此外,水培实验证明,OsADK1 可能参与植物对磷饥饿的响应。本研究发现了 OsPHR1/2/3 作为参与共生各个阶段的菌根相关基因的主调控因子的作用,并揭示了参与 AM 共生和植物 Pi 饥饿反应的新型植物类受体激酶(RLK)。
文献复现
此处对该文章的 RNA-seq 与 DAP-seq 数据分析部分进行复现,所得出的数据文件与代码均在 GitHub。
!!!注意:复现的 RNA-seq 分析中差异基因的数量以及 DAP-seq 实验中样品 peaks 的一致性与原文结果不同。
在与原文作者沟通后,发现这种差异可能是由于分析方法和过滤参数设置的不同(包括所使用的分析软件不同)造成的。原文可能应用了更为严格的筛选标准。不同的分析方法和筛选标准导致基因数量的差异是常见的现象。尽管如此,复现的结果仍然支持原文中的讨论。
RNA-seq
RNA-seq 数据在国家基因数据库:CNP0003633
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625460/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625461/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625462/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625463/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625464/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625465/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625466/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625467/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625468/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625469/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625470/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003633/CNS0625471/
下载的是 fastq.gz 文件,无需解压就可使用,将所有的文件放在 fastqgz 文件夹。
原文中描述The cleaned reads were mapped against the O. sativa ssp. Japonica cv Nipponbare reference genome (http://rice.plantbiology.msu.edu).并未提到使用什么软件进行比对,所以使用常用的 hisat2 来将 reads 回帖到参考基因组,使用 RGAP 数据库的基因组来建立 hisat2 索引,但是实际过程中,使用 RGAP 的基因组进行比对后,featureCounts 无法从 exon 进行计数,所以改用 Ensembl Plants 的基因组:
hisat2-build -p 4 Oryza_sativa.IRGSP-1.0.dna.toplevel.fa Oryza_sativa
先建立 files_list.txt 文件,将文件前缀写入,方便于后面对文件进行批处理:
Osphr123_-AM1
Osphr123_AM1
Osphr123_-AM2
Osphr123_AM2
Osphr123_-AM3
Osphr123_AM3
Wildtype_-AM1
Wildtype_AM1
Wildtype_-AM2
Wildtype_AM2
Wildtype_-AM3
Wildtype_AM3
将测序数据回帖到参考基因组:
cat ./files_list.txt | while read id;
do
hisat2 \
-x ./Oryza_sativa/Oryza_sativa \
-p 5 \
-1 ./fastqgz/${id}_1.fastq.gz \
-2 ./fastqgz/${id}_2.fastq.gz \
-S ./compared/${id}.sam
done
- -p 指定使用的线程数
- -x 指定参考基因组的索引文件的路径
- -1 指定测序数据的第一个配对端的文件路径
- -2 指定测序数据的第二个配对端的文件路径
- -S 指定输出比对结果的文件路径
完成后便得到了 sam 文件,接着需要将 sam 文件排序压缩为 bam 文件:
cat ./files_list.txt | while read id ;do
samtools \
sort \
-n -@ 5 \
./compared/${id}.sam \
-o ./sorted/${id}.bam
done
文中同样未提到使用的定量软件,于是使用常用的 featureCounts:
nohup featureCounts \
-T 5 \
-t exon \
-g Name \
-a Oryza_sativa.IRGSP-1.0.58.chr.gff3 \
-o counts \
-p Osphr123_-AM1.bam Osphr123_AM1.bam Osphr123_-AM2.bam Osphr123_AM2.bam Osphr123_-AM3.bam Osphr123_AM3.bam Wildtype_-AM1.bam Wildtype_AM1.bam Wildtype_-AM2.bam Wildtype_AM2.bam Wildtype_-AM3.bam Wildtype_AM3.bam &
- -T 指定使用的线程数
- -t 指定要计数的特征类型
- -g 指定用于分组的基因标识符
- -a 指定参考基因组的注释文件
- -o 指定输出特征计数的文件名
- -p 指定要计数的 BAM 文件
得到 counts 矩阵后,默认为转录本水平的表达矩阵,进行后续分析需要使用基因水平的表达矩阵,使用 TBtools 的 Trans Value Sum 工具将不同的转录本进行合并,随后进行 fpkm 标准化:
rm(list = ls())
counts <- read.csv(
'counts',
header = TRUE,
sep = '\t',
# row.names = "Geneid",
comment.char = '#',
check.names = FALSE
)
# 对第 6 列以后的数据进行 fpkm 计算
for (clm in colnames(counts)[6:ncol(counts)]) {
col_fpkm <- paste0(clm, "_FPKM") # 新列的名称,加上"_FPKM"后缀
total <- sum(counts[, clm]) # 计算每个样本的总读数
counts[col_fpkm] <- (counts[, clm] * 10^6) / (counts[, "Length"] / 1000) # 使用相应样本的长度值计算 FPKM 并添加 FPKM 列
}
# 删掉原有的 counts
counts = counts[,-c(2:19)]
numeric_mask <- sapply(counts, is.numeric)
counts[numeric_mask] <- lapply(counts[numeric_mask], function(x) ifelse(x < 1, x + 1, x))
write.table(counts, file = 'fpkm_output', sep = '\t', row.names = FALSE)
得到标准化的矩阵后,进行差异表达分析,原文The differentially expressed genes (DEGs) were identified using the R package DEGseq v.1.20.0 in different comparisons with P-value < 0.001 and |Fold change| > 2.提到使用 DEGseq 包进行,但是 DESeq2 包相对于 DEGseq 包功能更加强大、更精确和稳健,所以此处使用 DESeq2 包进行差异表达分析:
rm(list = ls())
Sys.setenv(LANGUAGE = "en")
library(DESeq2)
fpkm = read.csv(
'fpkm_merge',
header = T,
sep = '\t',
row.names = "Geneid",
comment.char = '#',
check.name = F
)
# 将小数四舍五入为整数
fpkm <- round(fpkm)
# 将空值全部写为 1
numeric_mask <- sapply(fpkm, is.numeric)
fpkm[numeric_mask] <- lapply(fpkm[numeric_mask], function(x) ifelse(is.numeric(x) & x < 1, x + 100, x))
# 保留行相加大于 10 的数据
fpkm <- fpkm[rowSums(fpkm)>10, ]
# 保证所有小数都转化为整数
fpkm[-1, ] <- apply(fpkm[-1, ], 2, as.integer)
# 如果有缺失值,删除这些行/列
missing_values <- sum(is.na(fpkm))
if (missing_values > 0) {
fpkm <- na.omit(fpkm)
}
samples = data.frame(
sampleID = c("Osphr123_-AM1", "Osphr123_-AM2", "Osphr123_-AM3", "Osphr123_AM1", "Osphr123_AM2", "Osphr123_AM3", "Wildtype_-AM1", "Wildtype_-AM2", "Wildtype_-AM3", "Wildtype_AM1", "Wildtype_AM2", "Wildtype_AM3"),
sample = c("Osphr123_reduce_AM", "Osphr123_reduce_AM", "Osphr123_reduce_AM", "Osphr123_add_AM", "Osphr123_add_AM", "Osphr123_add_AM", "Wildtype_reduce_AM", "Wildtype_reduce_AM", "Wildtype_reduce_AM", "Wildtype_add_AM", "Wildtype_add_AM", "Wildtype_add_AM")
)
rownames(samples) = samples$sampleID
samples$sample = factor(samples$sample, levels = c('Osphr123_reduce_AM', 'Osphr123_add_AM', 'Wildtype_reduce_AM', 'Wildtype_add_AM'))
dds = DESeqDataSetFromMatrix(countData = fpkm, colData = samples, design = ~sample)
dds_count <- DESeq(dds, fitType = 'mean', minReplicatesForReplace = 7, parallel = FALSE)
Osphr123_reduce_AM_vs_Osphr123_add_AM <- results(dds_count, contrast = c('sample', 'Osphr123_reduce_AM', 'Osphr123_add_AM'))
Wildtype_reduce_AM_vs_Wildtype_add_AM <- results(dds_count, contrast = c('sample', 'Wildtype_reduce_AM', 'Wildtype_add_AM'))
Wildtype_add_AM_vs_Osphr123_add_AM <- results(dds_count, contrast = c('sample', 'Wildtype_add_AM', 'Osphr123_add_AM'))
Wildtype_reduce_AM_vs_Osphr123_reduce_AM <- results(dds_count, contrast = c('sample', 'Wildtype_reduce_AM', 'Osphr123_reduce_AM'))
result1 <- data.frame(Osphr123_reduce_AM_vs_Osphr123_add_AM, stringsAsFactors = FALSE, check.names = FALSE)
result2 <- data.frame(Wildtype_reduce_AM_vs_Wildtype_add_AM, stringsAsFactors = FALSE, check.names = FALSE)
result1 <- data.frame(Wildtype_add_AM_vs_Osphr123_add_AM, stringsAsFactors = FALSE, check.names = FALSE)
result2 <- data.frame(Wildtype_reduce_AM_vs_Osphr123_reduce_AM, stringsAsFactors = FALSE, check.names = FALSE)
write.table(result1, 'Osphr123_reduce_AM_vs_Osphr123_add_AM.DESeq2.txt', col.names = NA, sep = '\t', quote = FALSE)
write.table(result2, 'Wildtype_reduce_AM_vs_Wildtype_add_AM.DESeq2.txt', col.names = NA, sep = '\t', quote = FALSE)
write.table(result1, 'Wildtype_add_AM_vs_Osphr123_add_AM.DESeq2.txt', col.names = NA, sep = '\t', quote = FALSE)
write.table(result2, 'Wildtype_reduce_AM_vs_Osphr123_reduce_AM.DESeq2.txt', col.names = NA, sep = '\t', quote = FALSE)
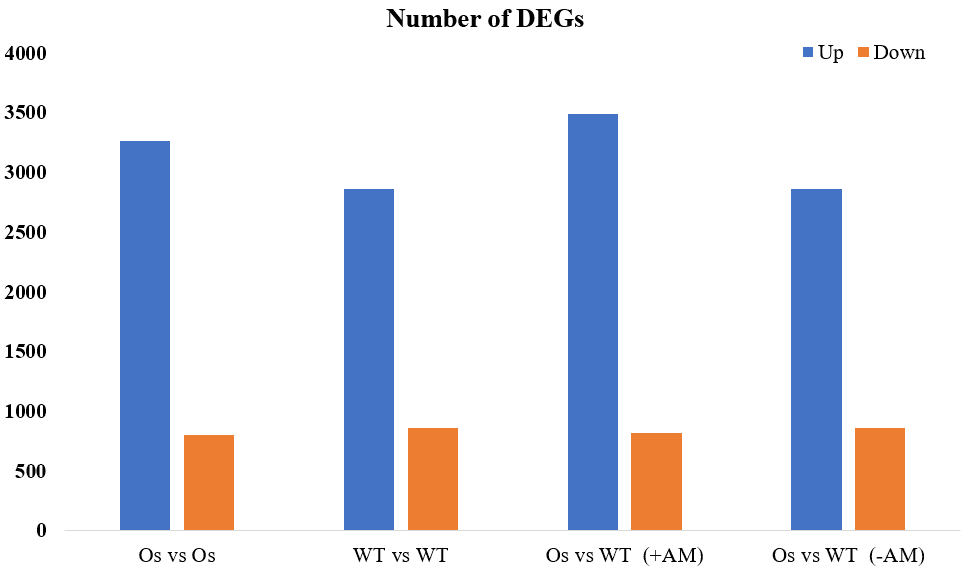
根据阈值对数据进行筛选:
| Up | Down | |
|---|---|---|
| Os vs Os | 3266 | 803 |
| WT vs WT | 2865 | 858 |
| Os vs WT (+AM) | 3489 | 816 |
| Os vs WT (-AM) | 2865 | 858 |
DAP-seq
DAP-seq 数据在国家基因数据库:CNP0003634
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003634/CNS0625472/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003634/CNS0625473/
wget -c -nH -np -r -R "index.html*" --cut-dirs 4 ftp://ftp.cngb.org/pub/CNSA/data5/CNP0003634/CNS0625474/
下载的是 fastq.gz 文件,无需解压就可使用,将所有的文件放在 fastqgz 文件夹。
根据原文the cleaned DAP-seq reads were mapped to the Nipponbare reference genome (https://rice.plantbiology.msu.edu/) using Bowtie2使用 RGAP 数据库的基因组来建立 bowtie2 索引,但是实际过程中,使用 RGAP 的基因组进行比对后,无法使用 samtools 建立索引,所以改用 Ensembl Plants 的基因组:
bowtie2-build Oryza_sativa.IRGSP-1.0.dna.toplevel.fa Oryza_sativa
先建立 files_list.txt 文件,将文件前缀写入,方便于后面对文件进行批处理:
GFP
OsPHR2_Rep1
OsPHR2_Rep2
将测序数据回帖到参考基因组:
mkdir compared
cat ./files_list.txt | while read id;
do
bowtie2 \
-p 10 -x Oryza_sativa/Oryza_sativa \
-1 ./fastqgz/${id}_1.fastq.gz \
-2 ./fastqgz/${id}_2.fastq.gz \
-S ./compared/${id}.sam
done
- -p 指定使用的线程数
- -x 指定参考基因组的索引文件的路径
- -1 指定测序数据的第一个配对端的文件路径
- -2 指定测序数据的第二个配对端的文件路径
- -S 指定输出比对结果的文件路径
完成后便得到了 sam 文件,接着需要将 sam 文件排序压缩为 bam 文件并为 bam 文件建立索引,以便进行进行后续的分析:
mkdir sorted
cat ./files_list.txt | while read id;
do
samtools sort ./compared/${id}.sam -o ./sorted/${id}.bam
samtools index ./sorted/${id}.bam
done
根据原文Peak calling was performed using the MACS2 with Q-value ≤ 0.05.使用 macs2 进行 calling peaks:
mkdir peaks
cat ./files_list.txt | while read id;
do
macs2 callpeak -t ./sorted/${id}.bam -f BAM -g 3.6e+8 -n ./peaks/${id} --qvalue 0.05
done
- -f 指定输入文件格式
- -g 指定基因组的大小
- –qvalue 指定 qvalue 的值
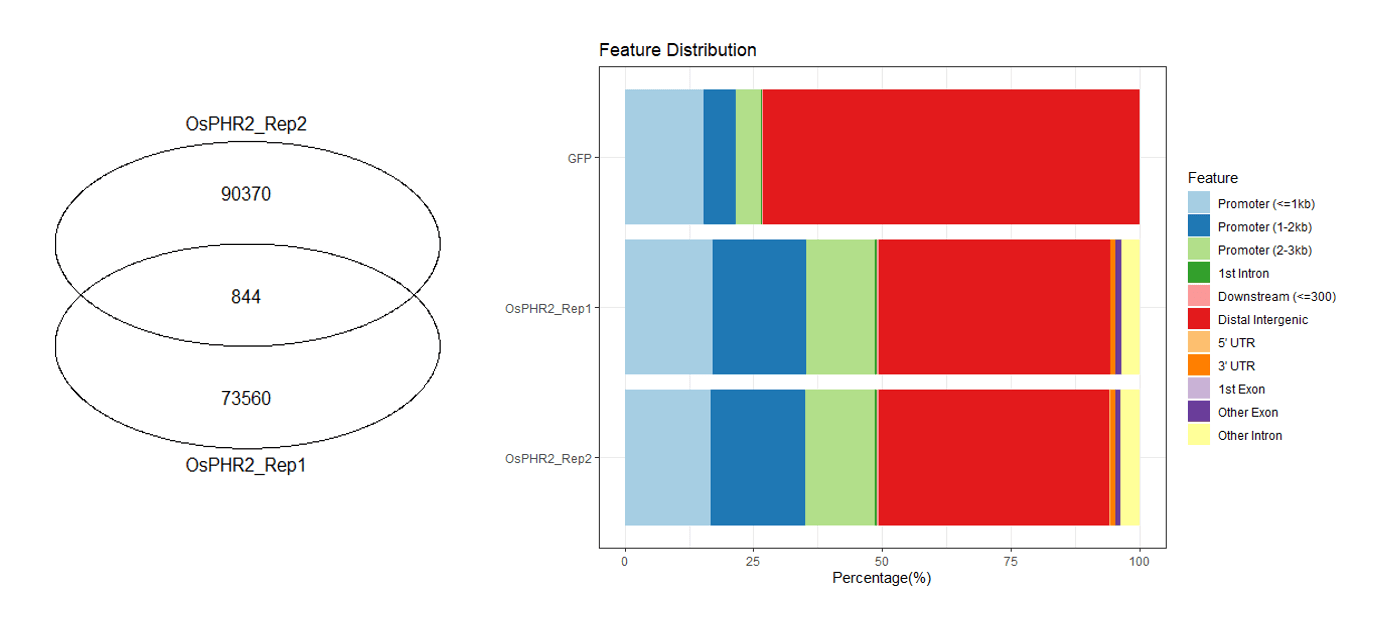
对 peaks 进行注释与可视化,由于原文并未提到使用的软件,所以使用常见的 ChIPseeker 包来进行,并且绘制韦恩图:
rm(list = ls())
library("GenomicFeatures")
library("ChIPseeker")
spombe <- makeTxDbFromGFF("./peaks/all.gff3")
gfp <- readPeakFile('./peaks/GFP.bed')
Rep1 <- readPeakFile('./peaks/OsPHR2_Rep1.bed')
Rep2 <- readPeakFile('./peaks/OsPHR2_Rep2.bed')
peaks <- list(GFP=gfp, OsPHR2_Rep1=Rep1, OsPHR2_Rep2=Rep2)
OsPHR2_peaks <- list(OsPHR2_Rep1=Rep1, OsPHR2_Rep2=Rep2)
peakAnno <- lapply(peaks, annotatePeak, tssRegion = c(-3000, 3000), TxDb = spombe)
plotAnnoBar(peakAnno)
vennplot(OsPHR2_peaks)
得到结果:
根据原文The coverage was generated by DeepTools, normalizing to RPM (Reads per million mapped reads) and then used for IGV view.The regions of the peaks were defined based on the annotations of the reference genome使用 deeptools 生成 bigWig 文件,由于现在的 deeptools 没有 RPM 这个标准化的格式,所以设置为 FPKM:
mkdir bigwig
cat ./files_list.txt | while read id;
do
bamCoverage -b ./sorted/${id}.bam -o ./bigwig/${id}.bw --normalizeUsing RPKM
done
将 bigWig 文件导入到 IGV 进行查看:
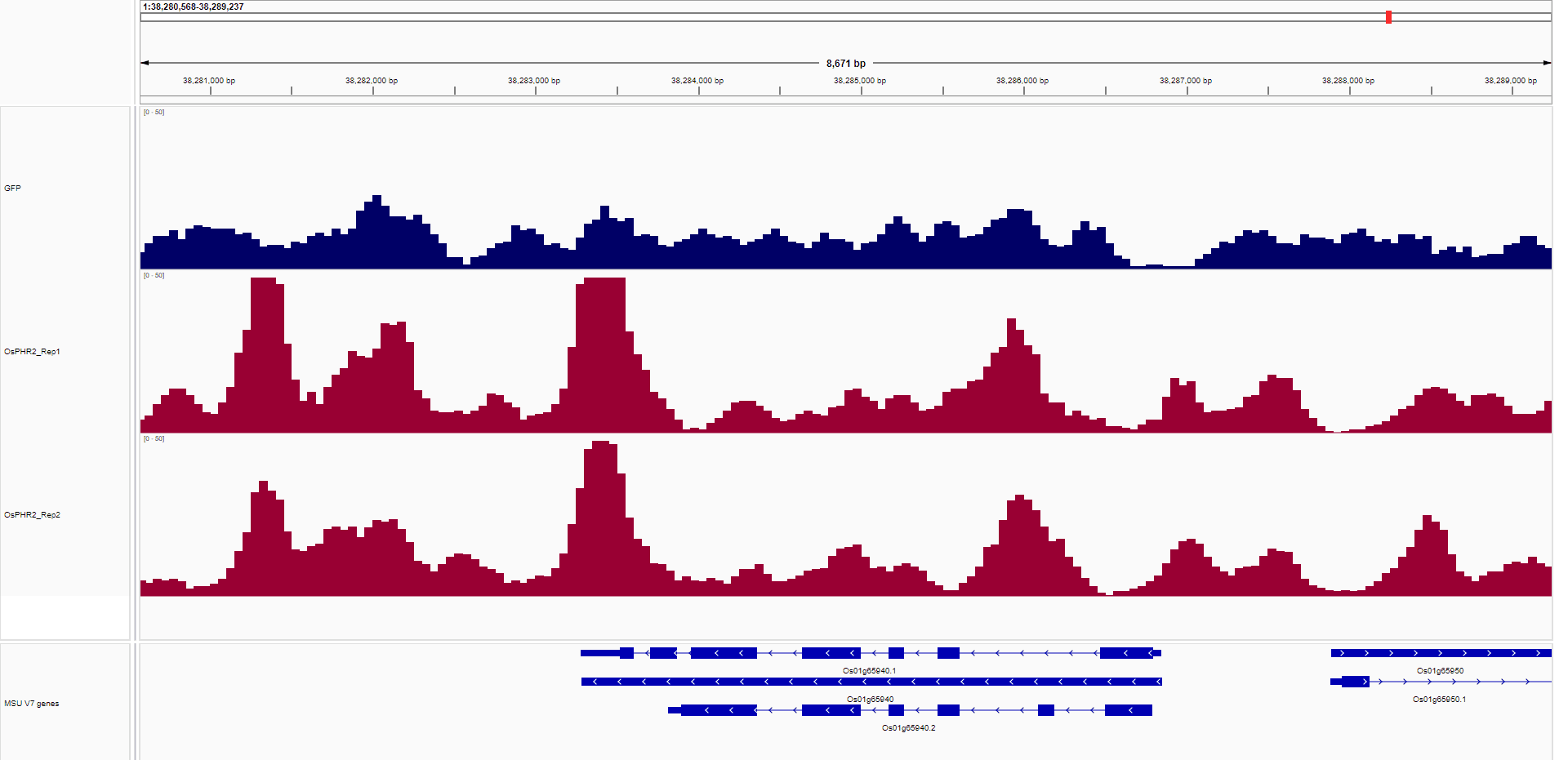
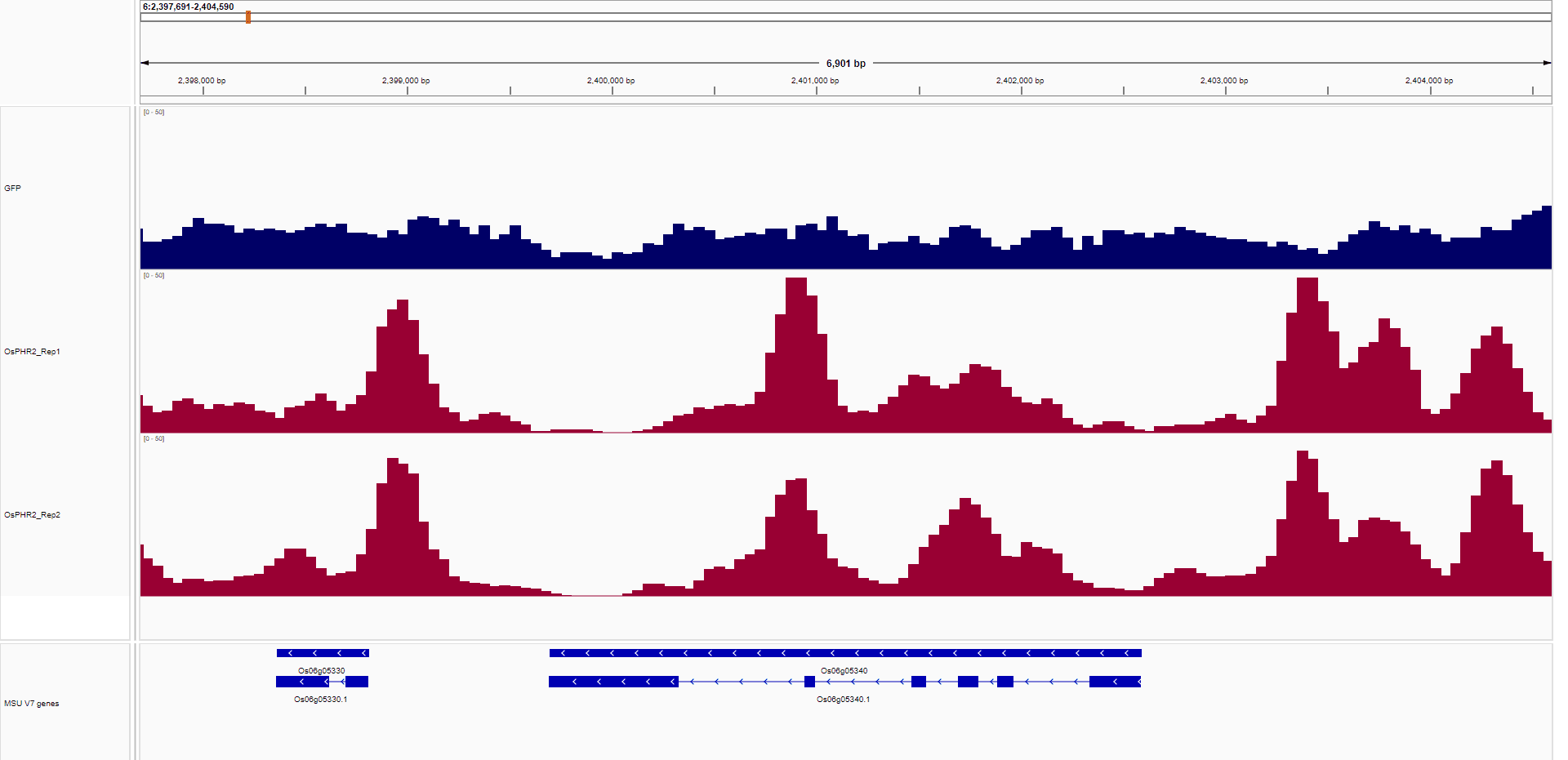
随后就要计算样本的信号丰度,这里需要一个 bed 文件,deeptools 文档说明可以从 UCSC 下载 bed 文件,但是 UCSC 数据库没有水稻的数据,因此根据 UCSC 上 BED format 的说明手动建立,从基因组注释文件中整理以下信息并保存为OsPHR2.bed文件:
chr7 14753753 14759288 OsPHR2 cds +
然后使用 computeMatrix 计算信号丰度:
cd bigwig
nohup computeMatrix \
scale-regions \
-p 10 \
-R OsPHR2.bed \
-S GFP.bw OsPHR2_Rep1.bw OsPHR2_Rep2.bw \
-b 3000 \
-a 3000 \
--regionBodyLength 5000 \
--skipZeros \
-o tss-tts.gz &
- scale-regions 将测量数据进行缩放以适应指定的区域进行计算
- -p 指定计算矩阵时使用的线程数
- -R 指定参考区域的 BED 文件
- -S 指定用于计算的信号文件
- -b 指定在参考区域的上游区域的长度
- -a 指定在参考区域的下游区域的长度
- -o 指定输出文件
- –regionBodyLength 指定在参考区域的区域主体长度
- –skipZeros 在计算矩阵时跳过值为零的区域
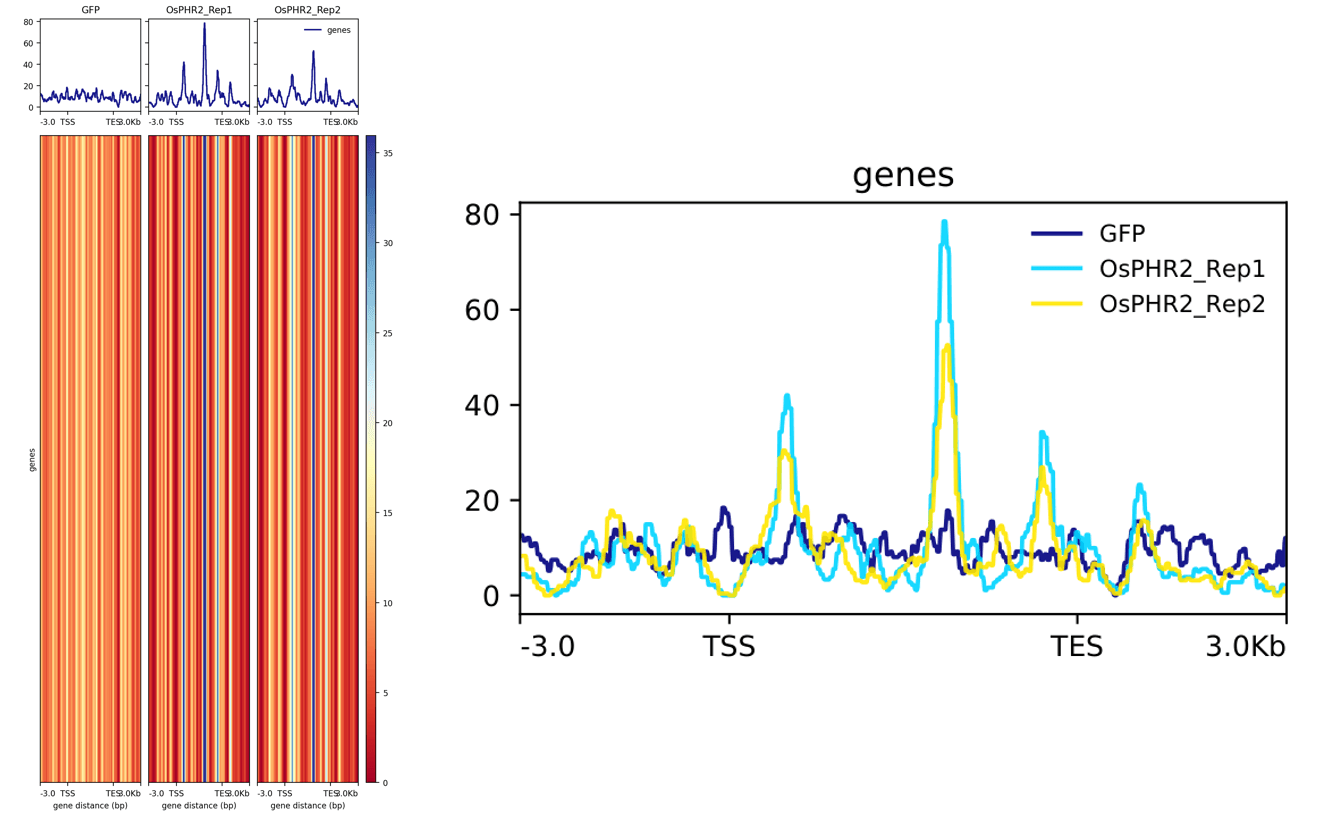
使用 plotProfile 和 plotHeatmap 进行可视化,plotProfile 将所有 bigWig 文件的结果绘制在同一个图中,而 plotHeatmap 将 bigWig 文件的结果分开绘制,并且绘制热图:
plotProfile --dpi 720 -m tss-tts.gz -out tss-tts.pdf --plotFileFormat pdf --perGroup
plotHeatmap -m tss-tts.gz -out tss-tts.png
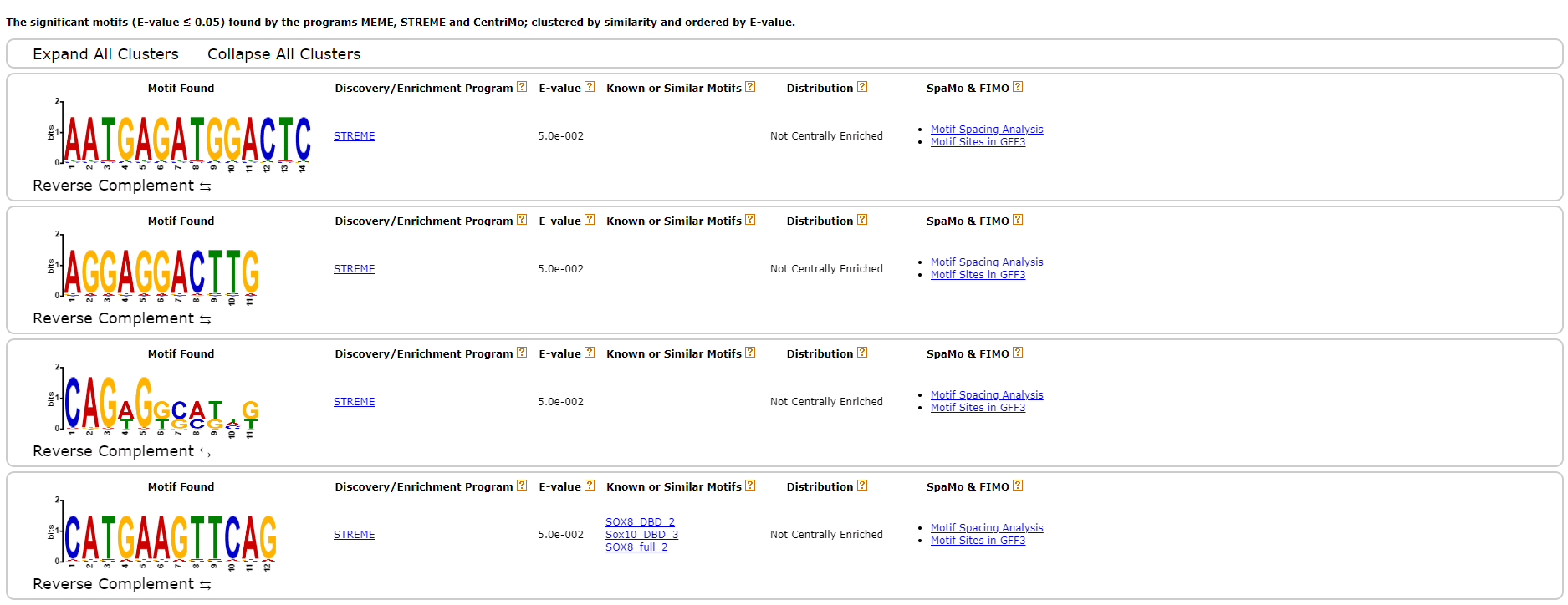
最后使用 MEME-Chip 工具来鉴定 OsPHR2 的 motif,使用 DNA 序列进行鉴定。