一次失败的分子动力学模拟
本文总阅读量 次
前不久,一位江苏大学的师兄请我帮忙做 gromacs 分子动力学模拟,他们找公司测得了酶的 pdb 蛋白结构,想喊我帮忙做 MD 模拟预测柔性位点,便于后期进行改造。研究了几天,还是失败了。
刚开始我是在超算上安装了 gromacs 准备进行,软件安装时遇到的报错是缺少 pocl 包,conda 安装上就可以了。
在生成拓扑文件时遇到问题:
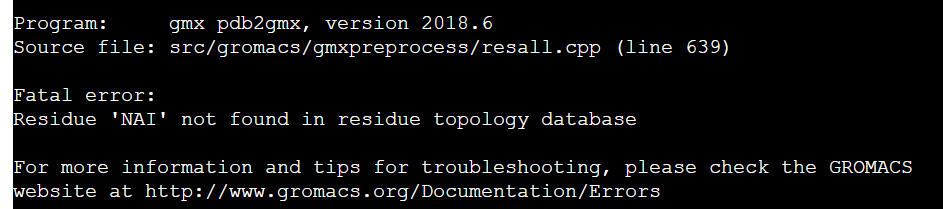
查了文档得知,这是因为 NAI 这个残基不在 gromacs 的标准残基库中,查了一些资料,参照 计算机化学公社 上的帖子打算用 acpype 来生成 gromacs 的拓扑文件,而不是用 gromacs 自带的 pdb2gmx,但还是报错,依旧是非标准残基导致的。
后来在 gromacs 社区查到可以自己手动补充非标准残基和力场到 gromacs 的配置中,于是参照官方文档和 Jerkwin 博客上的一篇 文章 打算手动补充,但是在创建残基的 rtp 文件时,做不下去了,不是我不想做,实在是这里需要的分子化学知识太多了,而且在创建包含残基的完整分子时,需要使用分子可视化软件手动截取一些残基的原子坐标并且进行封装,知识实在是跟不上了 …
只能暂时先放一放,后面找一下学校材化学院的老师请教一下。