JiaoYuan's Blog
共线性分析可以用 MCScanX 软件进行分析,但是这个软件的官网已经 404 了,而且利用命令行来进行分析比较麻烦,这里建议大家使用 TBtools 上的工具,这个软件里面集成了共线性分析的相关插件,并可以进行可视化操作,分析起来很方便。
概念
共线性是指同源基因在物种内或者物种之间的分布或排列关系。我们都知道,有相当多的基因在一个物种中不是以单拷贝的形式出现,而是多拷贝,具有基因序列重复事件,也就是说能行使相同功能的同源基因在同一个物种内可能不只有一条序列,而当我们想研究这些同源基因在同一个物种内的重复事件的时候,就是物种内的共线性分析。如果我们研究同源基因在不同物种内的分布情况,就是物种间的共线性分析,物种间可以是两个物种,也可以是多个物种。
原理
共线性分析最基本的原理是基于对基因序列进行 blast 比对,然后利用 MCScanX 软件对比对结果进行分析,这时候就需要有物种基因组的注释文件和比对结果,注释文件是用来分析这些基因所在的位置和数量,最后用可视化的软件把共线性分析的结果以图片形式展示出来。
所需工具及文件
TBtools 软件、物种基因组序列、注释文件
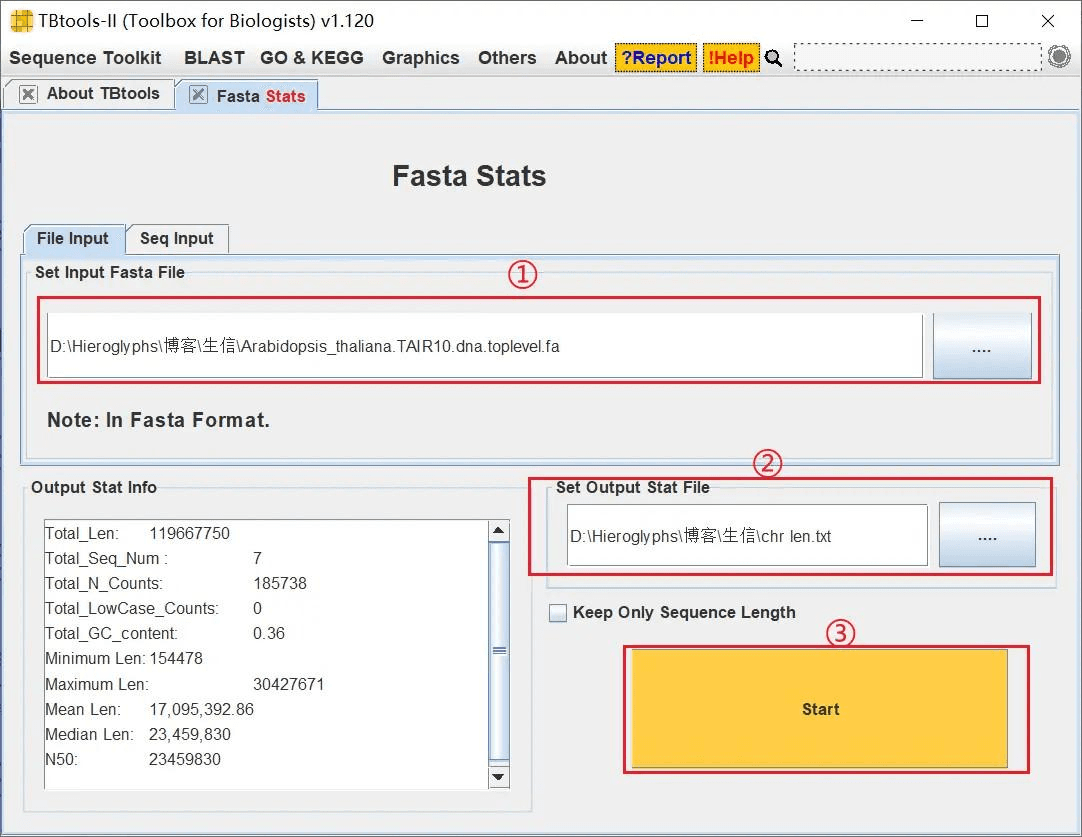
获取染色体长度文件
打开 TBtools 的 Fasta Stats 功能,导入物种基因组序列文件,设置好输出文件,获取染色体长度文件
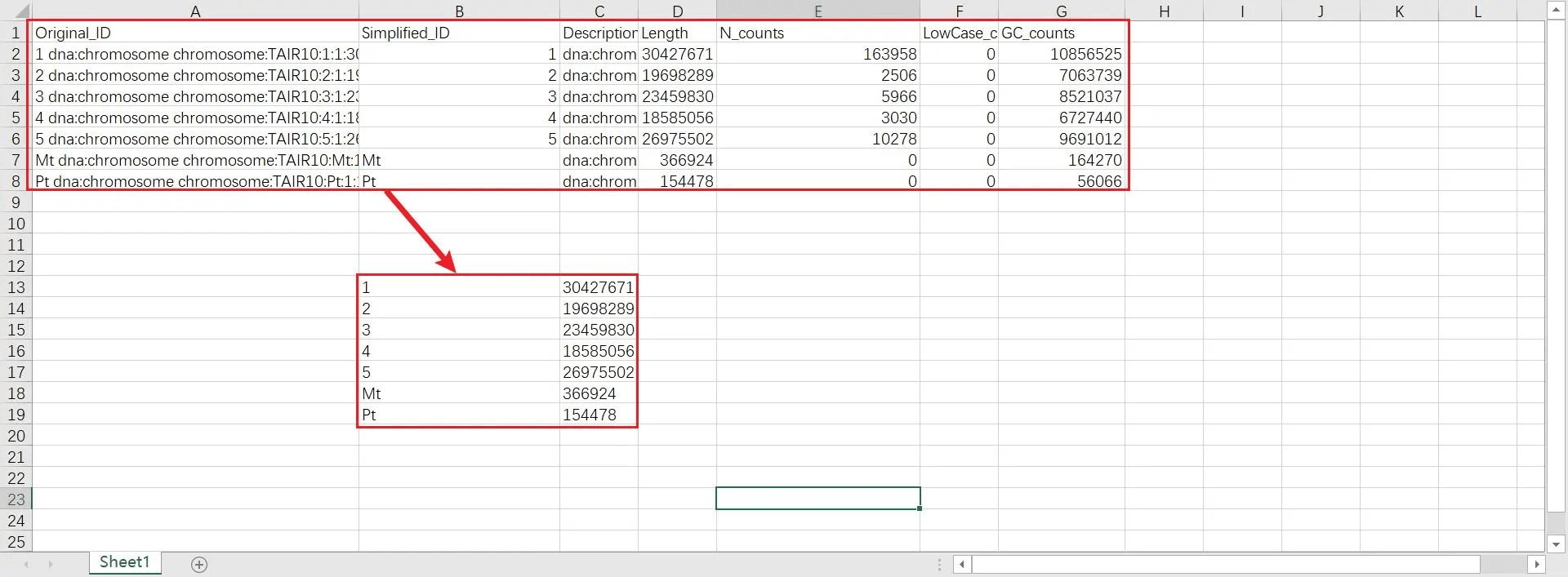
将 chr len.txt 文件的内容粘贴到 Excel,只保留 ID 和 length 两列的数据,删除 chr len.txt 文件中原有的数据,将 ID 和 length 两列数据保存在 chr len.txt 中
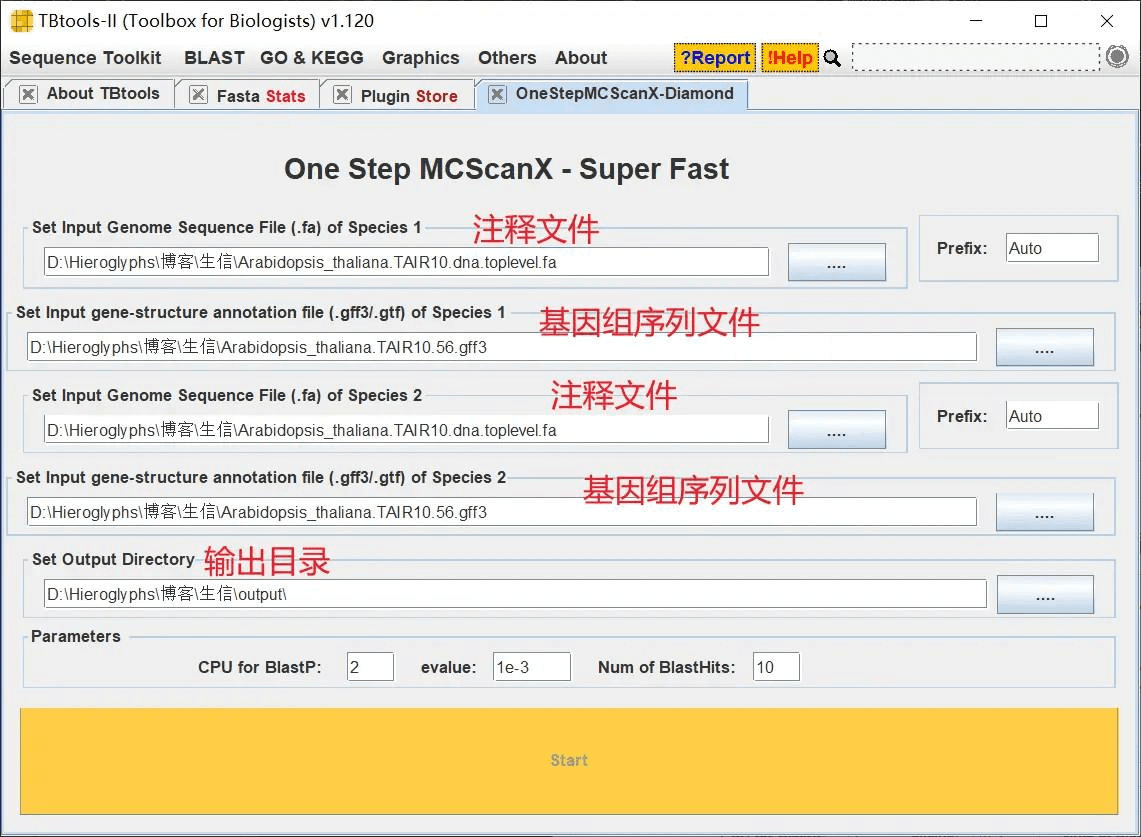
共线性分析
打开 TBtools 的 One Step MCScanX 功能,这里它需要输入两个物种的基因组序列文件和注释文件,我们两个都导入拟南芥的进行物种内的共线性分析,这个过程中如果出现报错,关掉即可,不用理会
获得关联文件
打开 TBtools 的 Text Merge for MCScanX 功能,导入上一步分析结果(在输出目录中)中的 Collinearity 文件,将分析结果转换为 GenePairTable
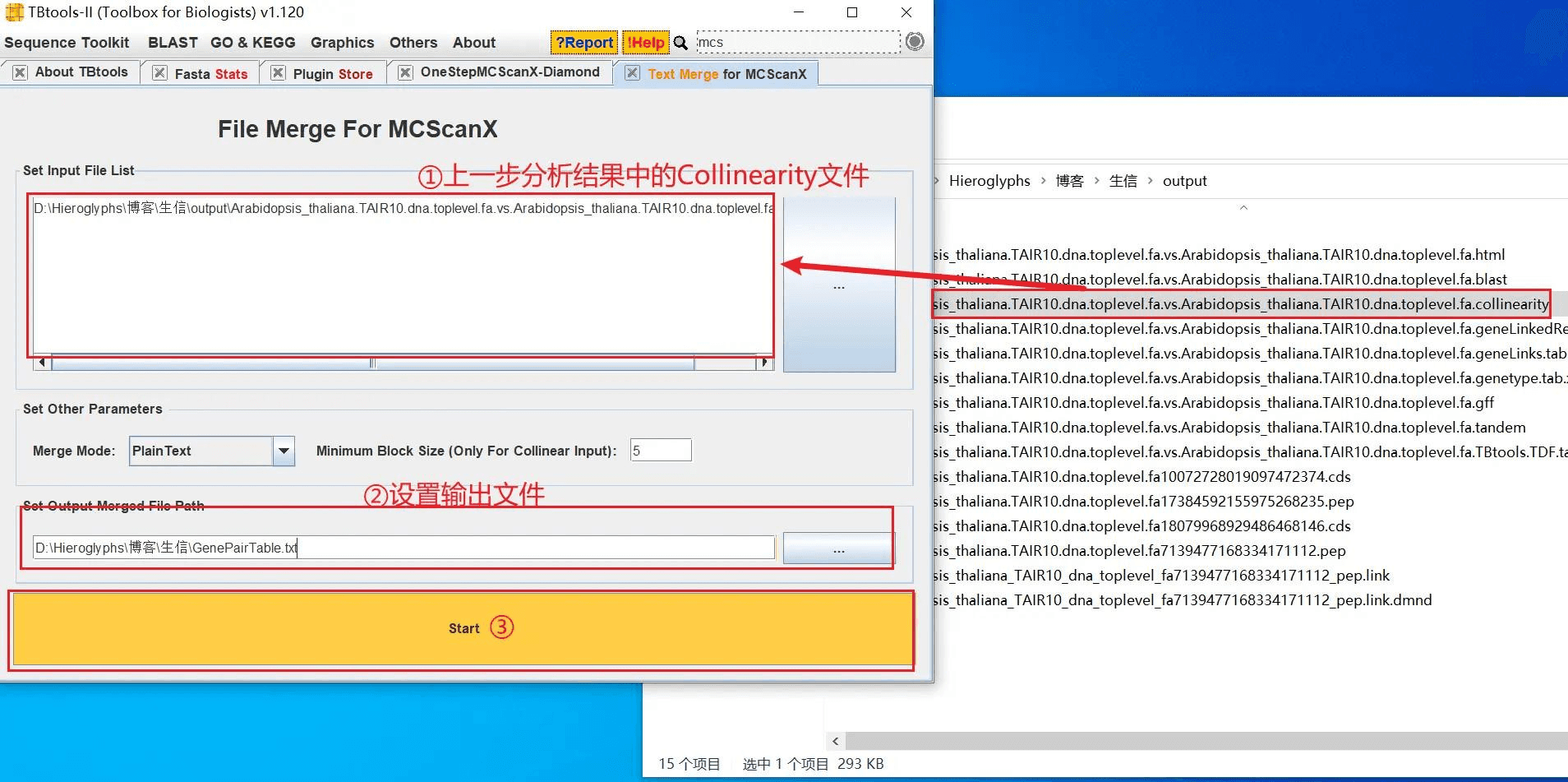
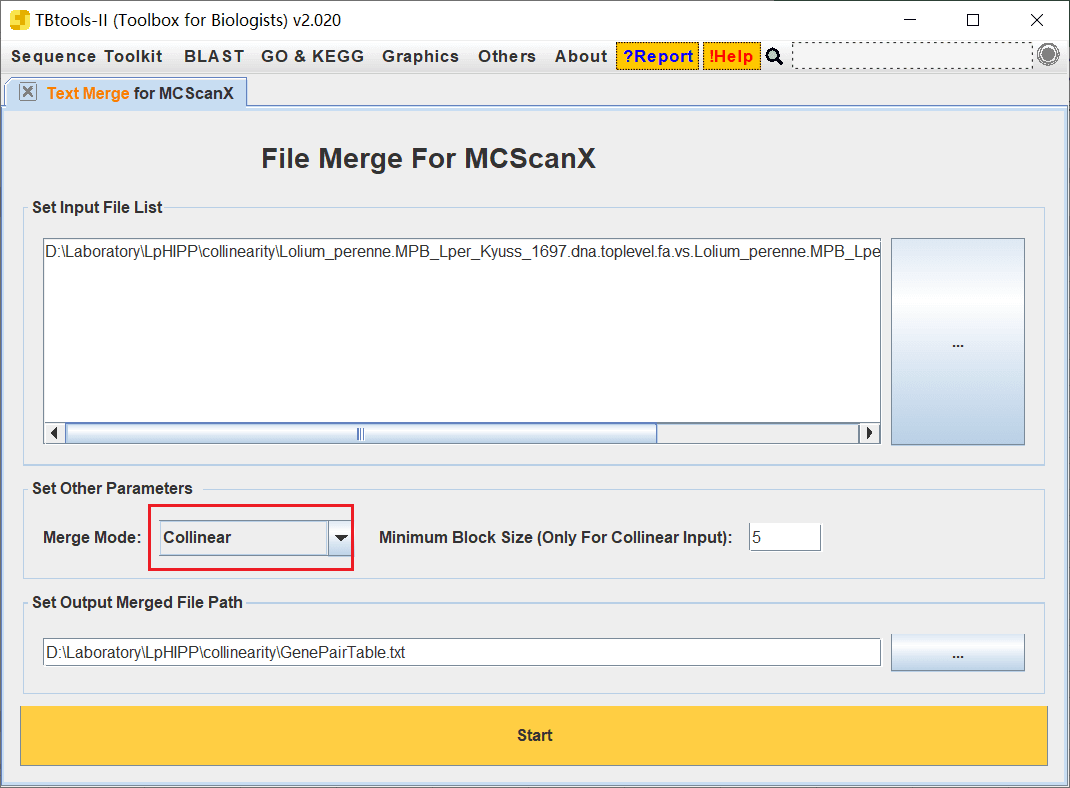
2023/11/14 补充,需要把合并模式改为 Collinear
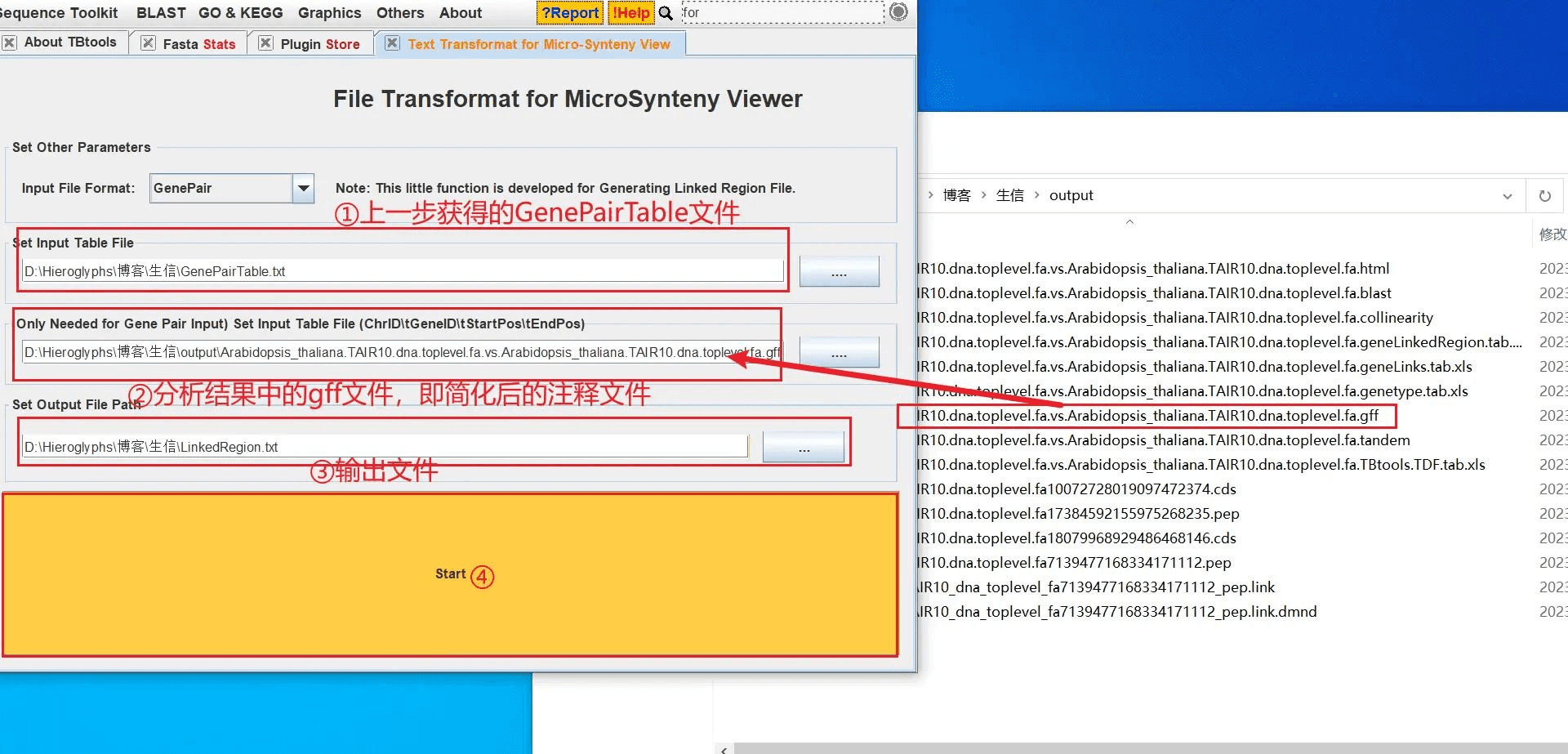
打开 TBtools 的 Text Transformat for Micro-Synteny View 功能,导入上一步获得的 GenePairTable 文件和分析结果中的以 .gff 为拓展名的文件(即简化后的注释文件),获得 LinkedRegion 文件,也就是基因间的关联文件 LinkedRegion
可视化
打开 TBtools 的 Advanced Circos 功能,依次导入 chr len.txt、GenePairTable.txt、LinkedRegion.txt 进行绘图
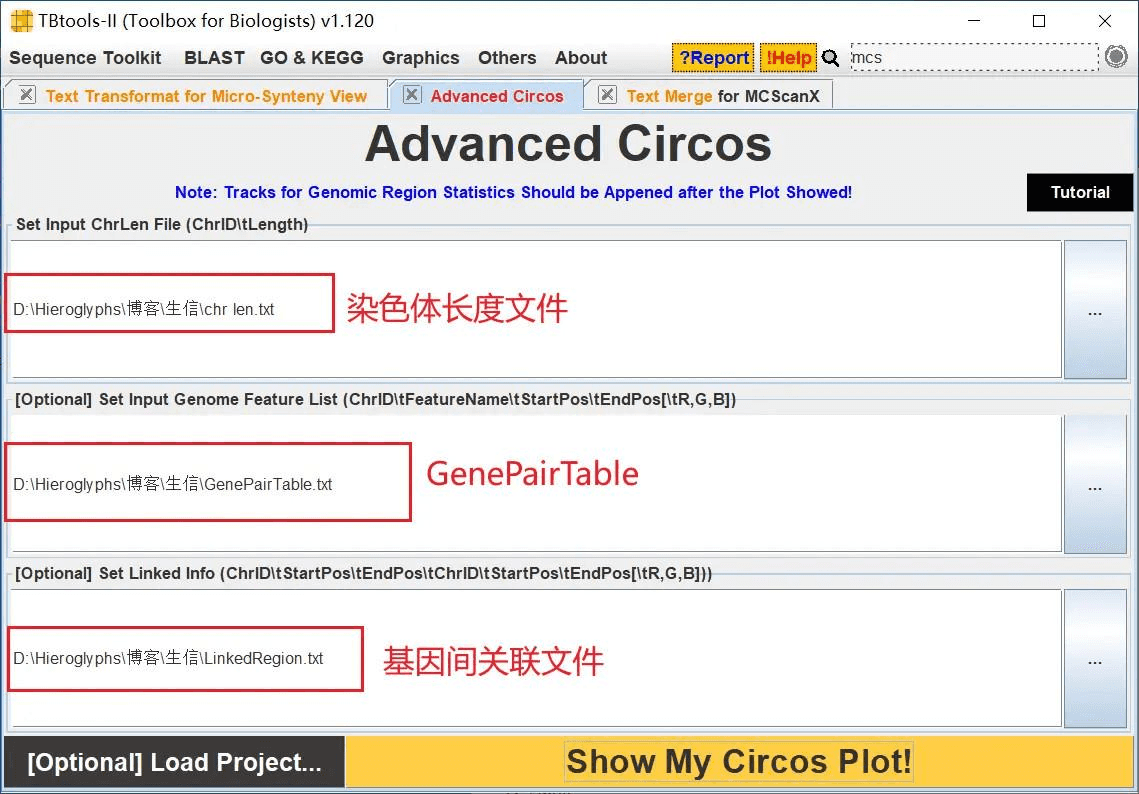
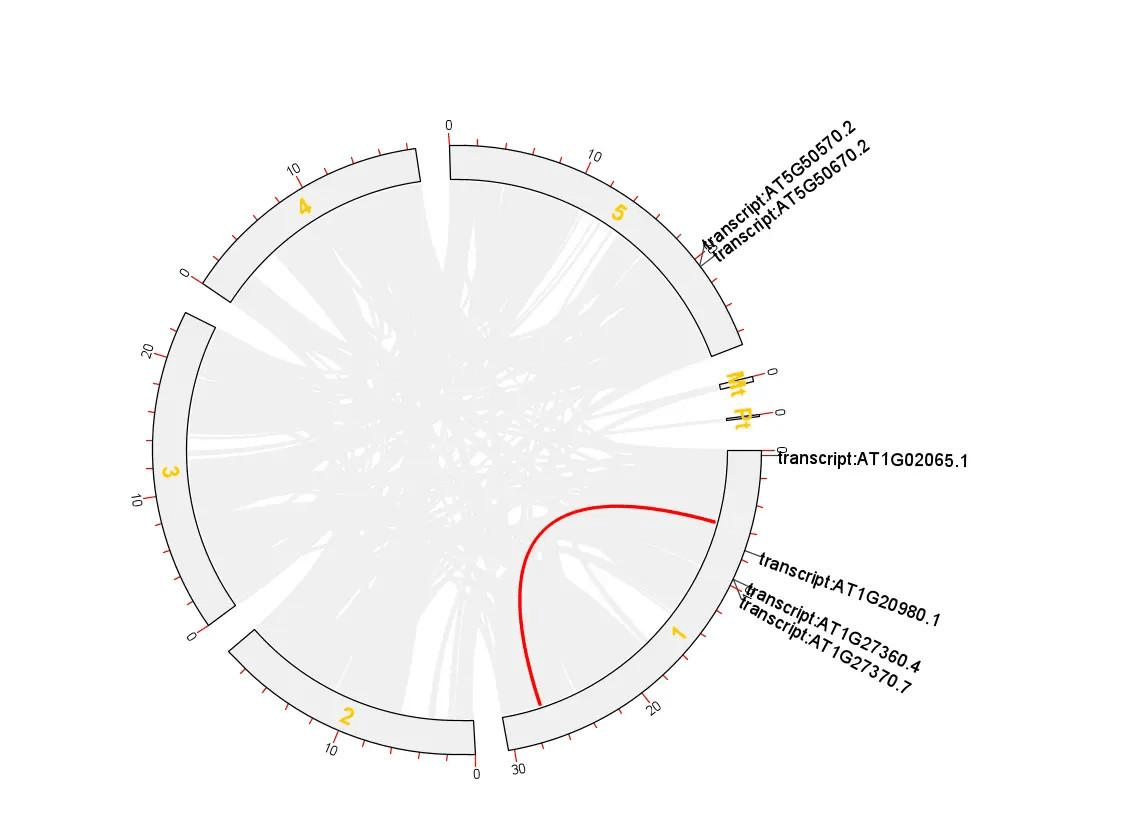
这样就得到拟南芥所有基因的共线性分析 Circos 图了
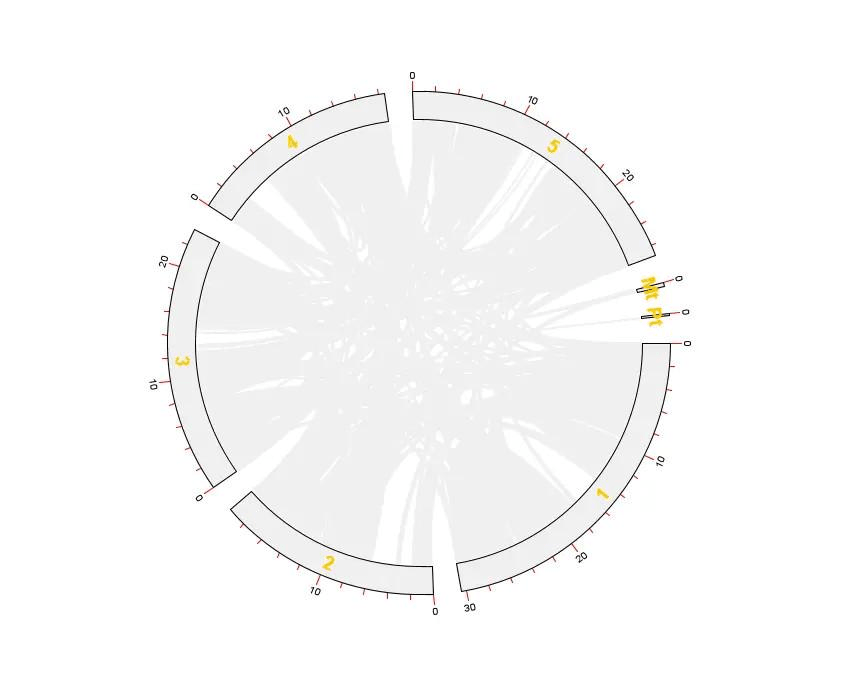
还没有结束
然而还没有结束,我们还需要突出显示出某些基因
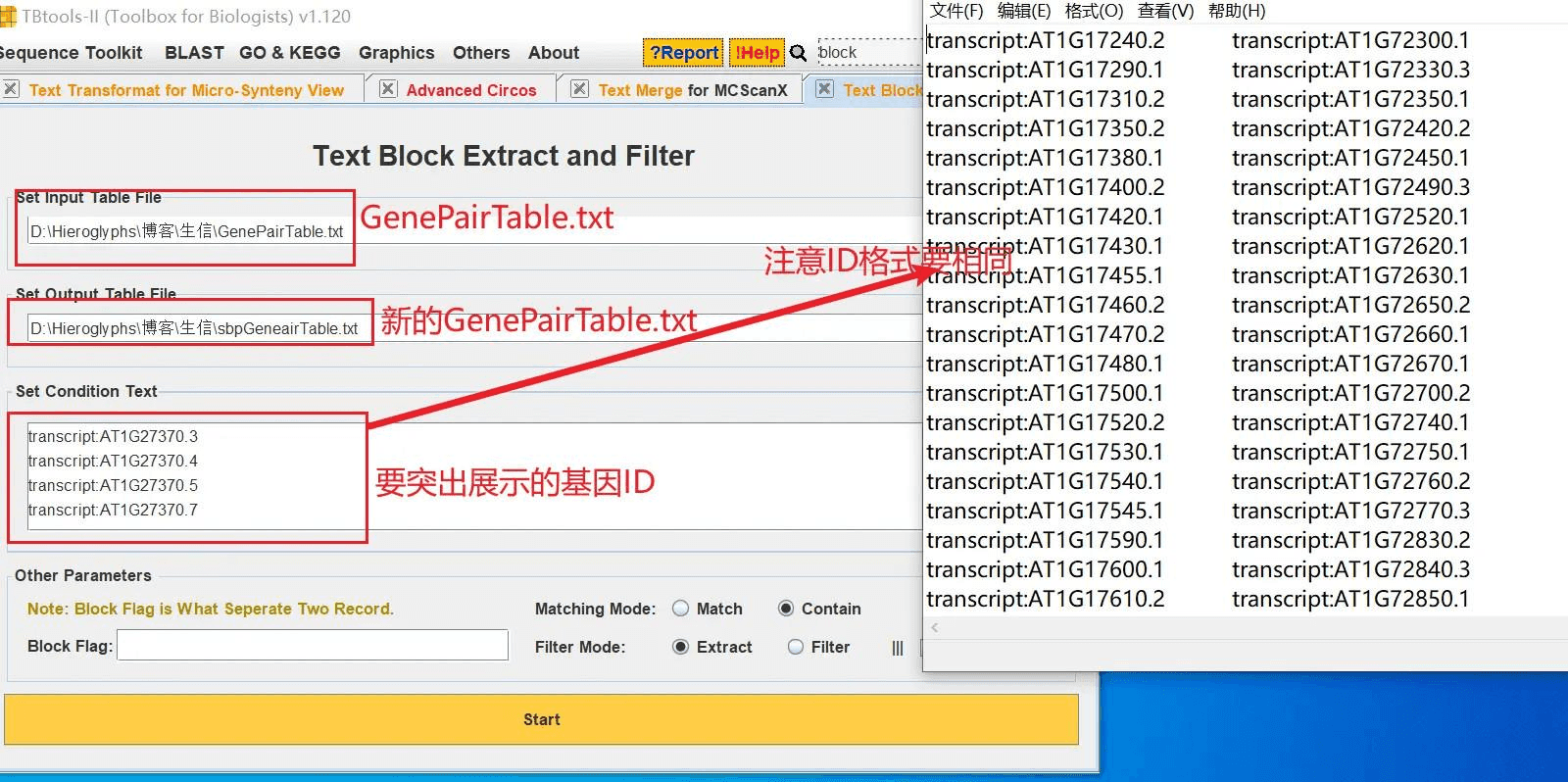
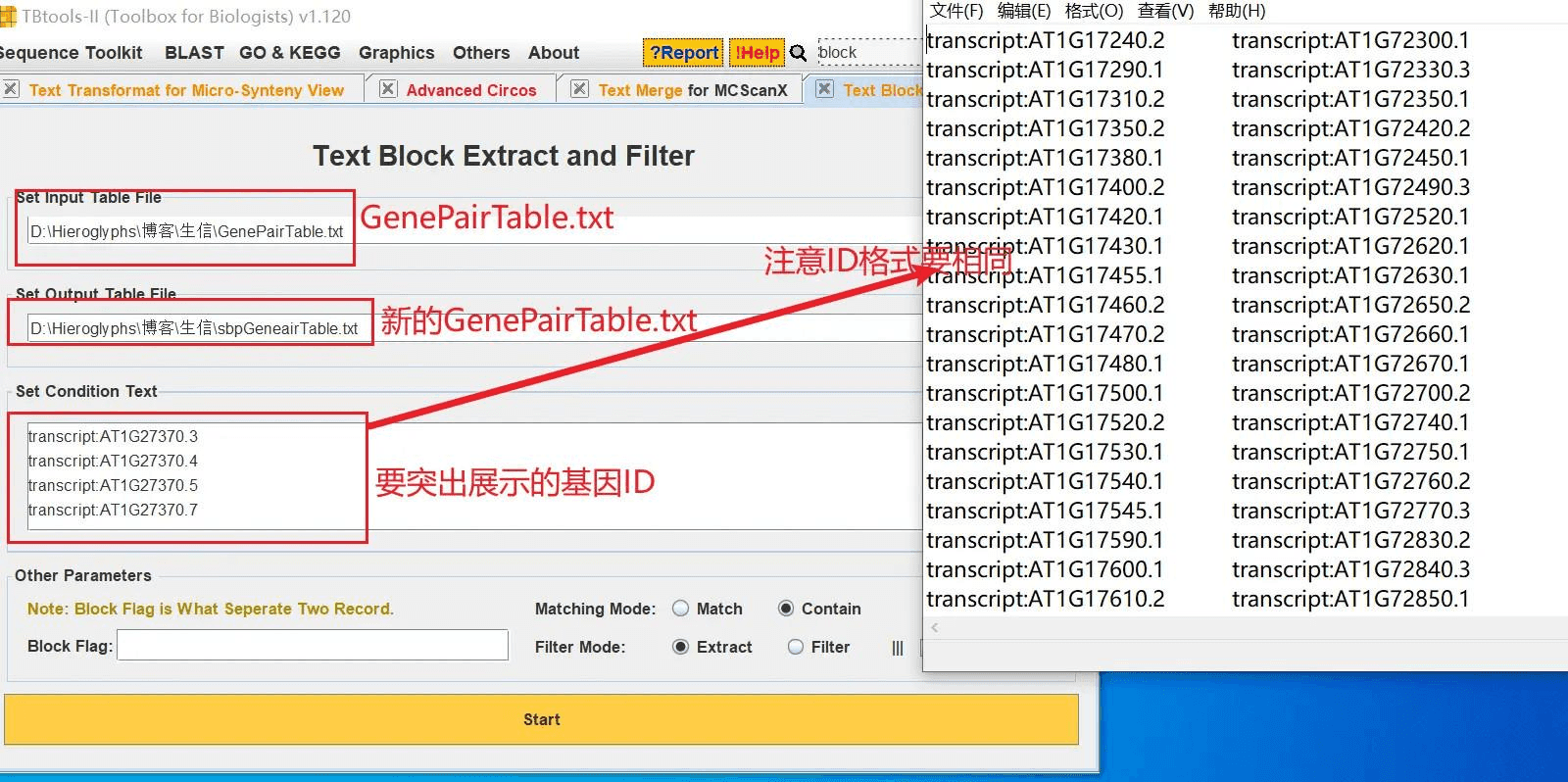
我随便抽取一些基因,打开 TBtools 的 Text Block Extract and Filter 功能,从 GenePairTable 文件中获得这些基因的对应关系,生成新的 GenePairTable 文件
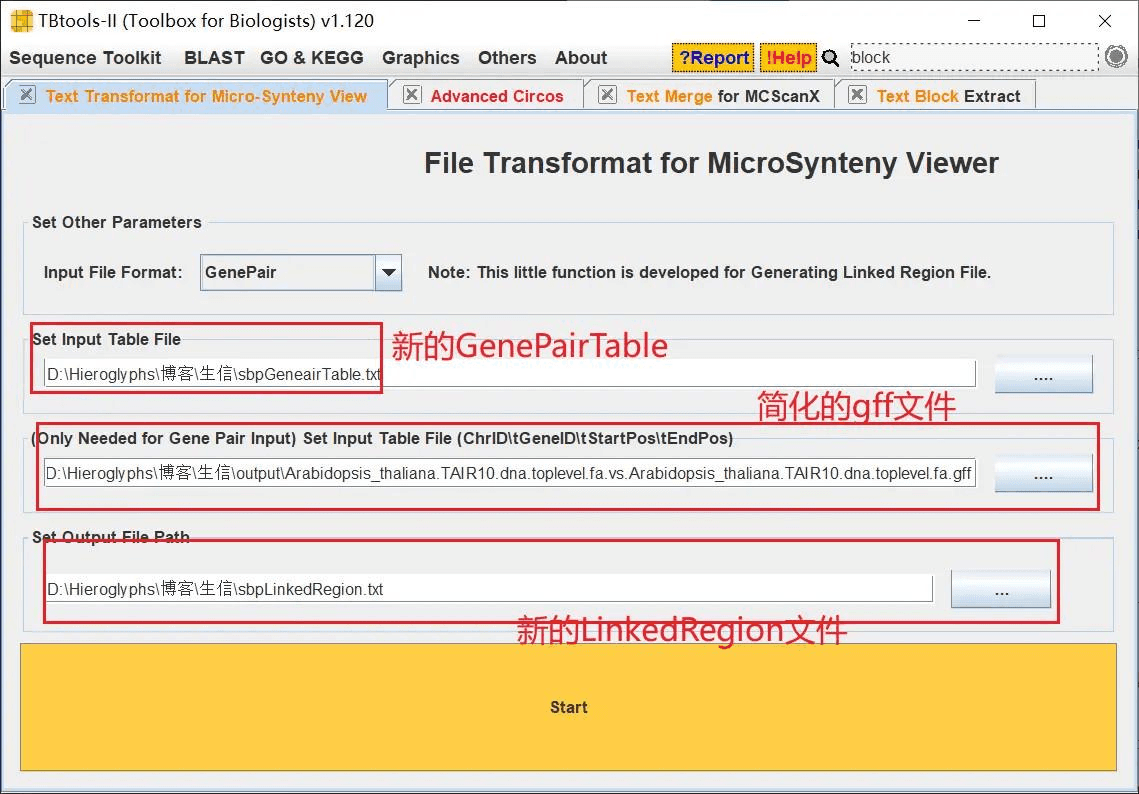
然后生成新的关联文件 LinkedRegion
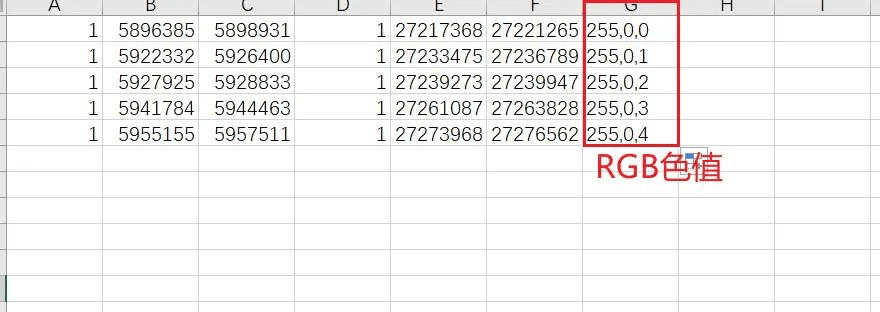
然后将新的 LinkedRegion.txt 文件的内容复制到 Excel 中,最后一列加上 RGB 色值
随后将 Excel 中的内容合并到之前的 LinkedRegion.txt 文件中或者新建一个文本文件保存,如果不想显示全部基因的共线性,仅仅显示选中的这些基因的共线性的话,可以直接使用刚刚保存的带有 RGB 色值的 LinkedRegion.txt 文件
2023/11/14 补充,可以使用 Text Merge for MCScanX 功能将两个文本合并为一个,合并模式使用 Plain Text
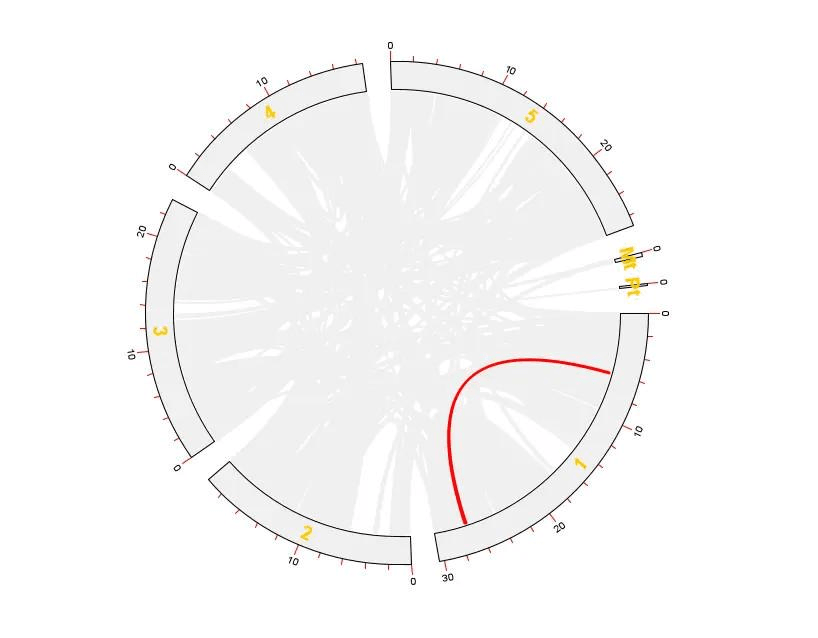
还差一些东西
我们似乎还需要显示出一些基因的名称,那就补充吧
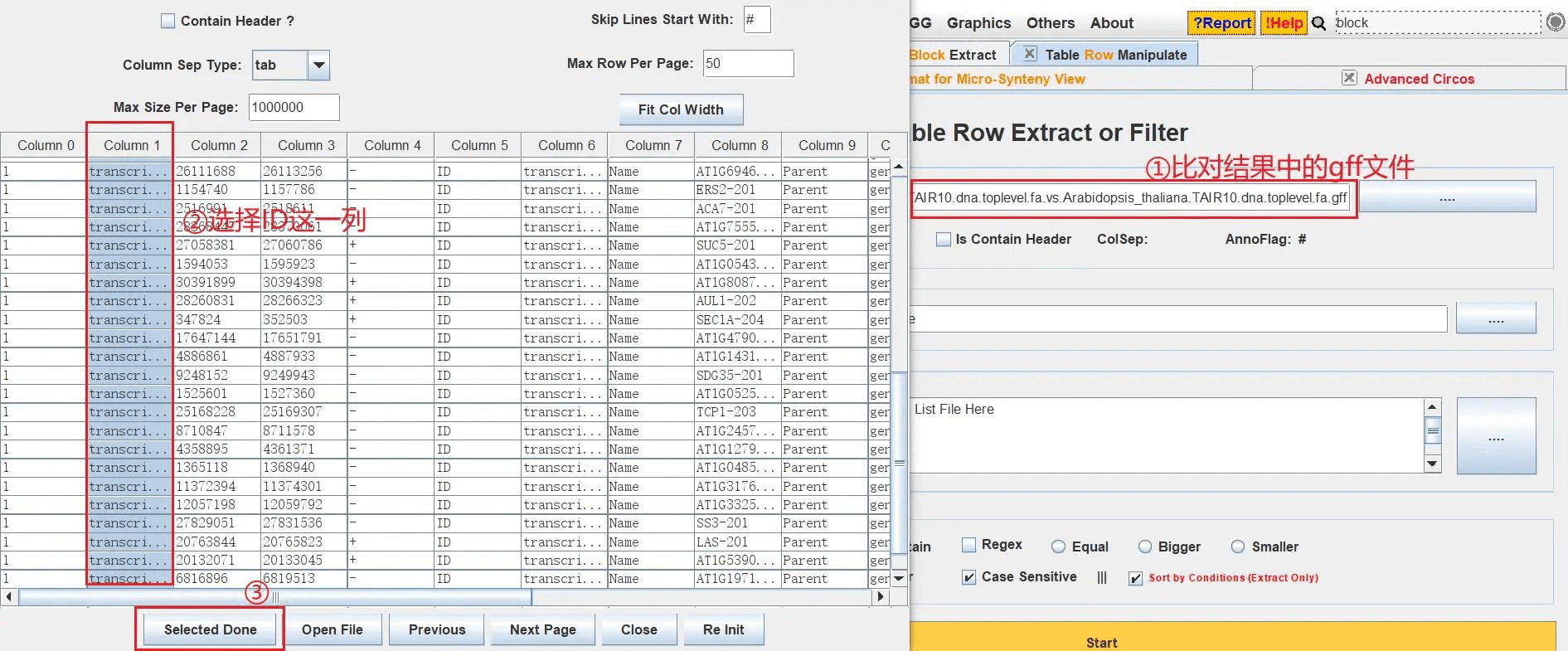
打开 TBtools 的 Table Row Extract or Filter 功能,导入分析结果中简化的注释文件,在弹出的窗口中选择 ID 这一列,在输入框中再输入要展示的基因的 ID(注意名称要对应),这里是需要生成一个用于展示 ID 的新的 GenePairTable 文件
最后依次输入染色体长度文件、展示 ID 的 GenePairTable 文件、带有 RGB 色值的 LinkedRegion.txt 文件 绘图即可
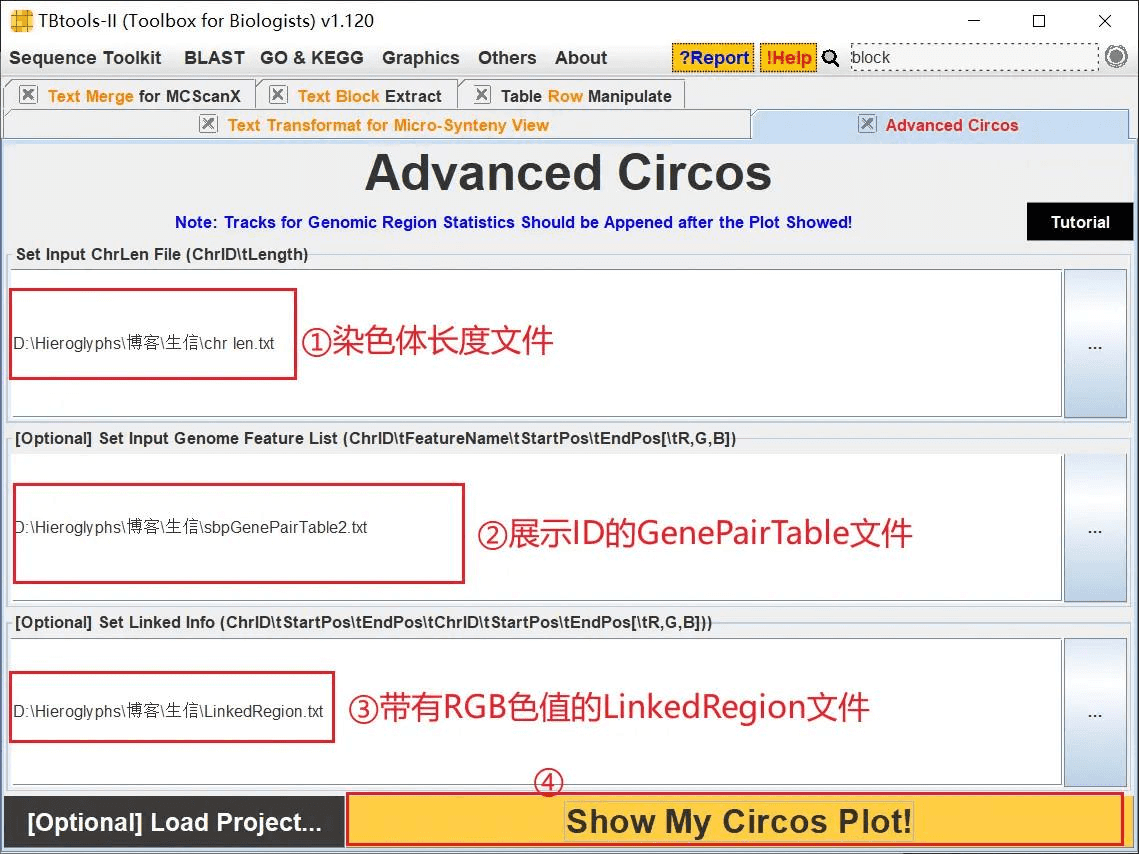
最终结果